|
|
|

Sinopsis: Síntesis y reactividad de compuestos orgánicos
|
Se estudia la síntesis y reactividad de todos los principales compuestos orgánicos, atendiendo los grupos funcionales más usuales
Agregado: 01 de MAYO de 2010 (Por Wilbert Rivera Muñoz) | Palabras: 15303 | Votar |
1 voto  | Promedio: 10
| Promedio: 10
|
Sin comentarios | Agregar Comentario | Promedio: 10
Categoría: Apuntes y Monografías > Química >
Material educativo de Alipso relacionado con Sinopsis Síntesis reactividad compuestos orgánicos
Enlaces externos relacionados con Sinopsis Síntesis reactividad compuestos orgánicos
Autor: Wilbert Rivera Muñoz (wlbrtrivera@gmail.com)
 Este apunte fue enviado por su autor en formato DOC (Word).
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. Este apunte fue enviado por su autor en formato DOC (Word).
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. |
UNIVERSIDAD AUTóNOMA TOMÁS FRíAS"
FACULTAD DE CIENCIAS PURAS
Carrera de Química

SíNTESIS Y REACTIVIDAD DE LOS COMPUESTOS ORGÁNICOS
|
|


Wilbert Rivera Muñoz
2006
PRESENTACIóN
La elaboración de una estrategia de síntesis, para una molécula determinada, demanda del químico un dominio de un conjunto de reacciones, que han sido probadas en el laboratorio de química, respecto de sus bondades, relacionadas, a sus altos rendimientos, la formación regioespecífica o en última instancia, de tener la certeza de que la estructura de interés podrá ser separada fácilmente, por métodos asequibles en el laboratorio, por ejemplo, destilación fraccionada, cromatografía de columna, etc.
Es posible que la presente sinopsis sobre la síntesis y reacciones de los principales compuestos orgánicos no abarquen todas las reacciones difundidas en la extensa literatura de química orgánica, sin embargo el hecho de que sea la primera publicación, en algún sentido le da al autor cierta tranquilidad, ya que conjuntamente los estudiantes, en un tiempo breve, se podrá dar a luz una obra mucho más completa y comentada,
En tal sentido, espero que la presente sea de utilidad y llene los requerimientos de aquellos espíritus investigadores de los estudiantes, que saben muy bien que lo que abarquen en clases nunca será suficiente.
Ante ellos asumo el compromiso de enriquecer la presente sinopsis en las próximas ediciones.
Wilbert Rivera Muñoz
Julio 2006
Potosí - Bolivia
CONTENIDO
Pág. ALCANOS Y CICLOALCANOS ALQUENOS Y CICLOALQUENOS ALQUINOS DERIVADOS HALOGENADOS ALIFÁTICOS ALCOHOLES Reacciones de los glicoles .... 23 ORGANOMETÁLICOS ÉTERES Y EPóXIDOS EPóXIDOS BENCENO ARENOS FENOLES HALUROS DE ARILO ALDEHIDOS Y CETONAS |
Pág. NITRILOS AMINAS Y SALES DE DIAZONIO SALES DE ARIL DIAZONIO ÁCIDOS CARBOXíLICOS DERIVADOS DE LOS ÁCIDOS CARBOXíLICOS Métodos de preparación............... 68 Reactividad ............................. 69 ÉSTERES Métodos de preparación ............. 70 Reactividad ............................ 71 AMIDAS Métodos de preparación .............. 72 Reactividad ............................. 73 ANHíDRIDOS Métodos de preparación .............. 74 Reactividad ............................ 74 ADICIONES Y CONDENSACIONES DE LOS ENOLES E IONES ENOLATO Formación y reactividad ............ 78 COMPUESTOS β- DICARBONíLICOS Formación y reactividad .............. 79 COMPUESTOS DE CARBONILO α,β- INSATURADOS Formación y reactividad ....... .... 81 OTRAS REACCIONES DE INTÉRES .................. 83 BIBLIOGRAFíA. |
ALCANOS Y CICLOALCANOS
Métodos de preparación de alcanos y cicloalcanos.
El petróleo es una mezcla compleja de hidrocarburos, la mayoría de los cuales son alcanos y cicloalcanos, y constituye el resultado final de la descomposición de la materia animal y vegetal sepultada bajo la corteza terrestre durante períodos muy largos, este será conjuntamente el gas natural, la principal fuente de obtención de los alcanos. Sin embargo, las reacciones que se señalan son otra alternativa de obtención en el laboratorio.
No |
REACCIóN |
EJEMPLOS |
1 |
Reducción de haluros de alquilo. |
|
2 |
Reducción de haluros de alquilo vía organometálicos |
|
3 |
Hidrogenación catalítica de alquenos, alquinos e hidrocarburos bencénicos o insaturados |
|
4 |
Síntesis de Corey-House. Reacciones de acoplamiento. |
|
5 |
Reacción de copulación de Wurtz (bajos rendimientos) |
|
6 |
Reacciones de ciclización. Las reacciones de eliminación de los dihalogenuros 1, 3 |
|
7 |
Las reacciones pericíclicas de los alquenos |
|
8 |
La adición de carbenos a olefinas |
|
9 |
Reducción de Clemmensen: Reducción de un aldehído o cetona en medio ácido |
|
10 |
Reducción de Wolff-Kishner: Reducción de un aldehído o cetona en medio básico |
|
11 |
Reacción de Diels- Alder seguida de reducción |
|
12 |
Síntesis electrolítica de Kolbe. Se somete a electrólisis la sal sódica del ácido, se preparan alcanos con un número par de átomos de carbono. |
|













Reacciones de los alcanos y cicloalcanos
Los alcanos y cicloalcanos debido a la falta de un "grupo funcional" no son muy reactivos, razón por la cual participan de pocas reacciones comparativamente con el resto de los compuestos orgánicos.
No |
REACCIóN |
EJEMPLOS |
13 |
Halogenación. |
|
14 |
Inserción de metileno. |
|
15 |
Isomerización |
|
16 |
Nitración En los hidrocarburos más complejos se pueden romper la cadena de carbono, dando resultados de todos los |
isómeros posibles. |
17 |
Reacciones de adición a cicloalcanos altamente tensionados (como por ejemplo el ciclopropano y ciclobutano) |
|
18 |
La cloración de alcanos con SO2Cl2.(cloruro de sulfurilo) La reacción ocurre en presencia de peróxidos y sólo los cicloalcanos producen derivados monoclorados con alto rendimiento: |
|
19 |
Deshidrogenación catalítica de cicloalcanos |
|
20 |
Reacción de Reed |
|
21 |
La oxidación de alcanos frecuentemente forma una mezcla de productos |
|









Métodos de preparación de alquenos y ciclcoalquenos.
Los métodos más comunes comprenden la eliminación de una molécula pequeña o simple, considerada inorgánica (ej. agua o haluro de hidrógeno) a partir de un compuesto saturado.
En muchos casos la eliminación sigue dos cursos diferentes, originándose los isómeros respectivos, para lo cual deben tomarse en cuenta reglas generales como la de Saytzeff y Markovnikov, o simplemente manejar con� sumo cuidado las condiciones de la reacción.
La reacción de Wittig tiene la ventaja de que la posición del doble enlace se puede predecir y conocer con certeza así como la reducción parcial de los alquinos.
No |
REACCIóN |
EJEMPLOS |
22 |
Deshidrogenación de compuestos alifáticos y arenos Cracking de alcanos: |
|
23 |
Deshidrohalogenación de haluros de alquilo y cicloalquilo. Se obtiene el alqueno más estable, Saytzeff, o sea el que tiene el doble enlace entre los C más sustituidos. Los H y X que se eliminan se hallan en la posición anti. Se obtiene la mezcla cis/trans. Bases típicas: KOH, C2H5ONa, (CH3)COK, etc. Facilidad de deshidrohalogenación de los RX: 3�>2�>1�. |
|
24 |
Deshalogenación de vec-dibromuros |
|
25 |
Deshidratación de Alcoholes Ácidos típicos: sulfúrico, fosfórico (H3PO4) y calor En algunos alcoholes existe transposición de hidruro y alquilo, dependiendo de la estructura del carbocatión Desplazamiento 1,2 de hidruro Desplazamiento 1,2- de alquilo Preparación de alcadienos: |
|
26 |
Otros procesos que nos permiten el mismo resultado: Facilidad de deshidratación de ROH: 3�>2�>1�. Estabilidad de carbocationes: 3�>2�>1�>metilo. |
|
27 |
Hidrogenación de alquinos Facilidad de formación de alquenos. R2C=CR2 > R2C=CHR > R2C=CH2 > RHC=CHR > RHC=CH2 |
|
28 |
Pirólisis de ésteres De este modo se evita las reacciones de transposición, de ahí que a veces los alcohol se esterifican previamente para formar una alqueno e su esqueleto carbonado |
|
29 |
Pirólisis de hidróxidos cuaternarios de amonio: Este tipo de pirólisis solo puede ocurrir si uno de los grupos alquilo unidos al nitrógeno tiene átomos de hidrógeno en el carbono beta. Se forma siempre el alqueno menos sustituido. Esta reacción se conoce como la reacción de degradación de Hofmann. |
|
30 |
La reacción de Wittig Se calienta aldehído o cetona con un alquilidén fosforano: (Ph)3P=CHR |
|
31 |
Reacciones Pericíclicas Reacciones de cicloadición: (reacciones de Diels - Alder) �R : debe ser un buen grupo atractor de electrones Reacciones electrocíclicas: Es una ciclación concertada a partir de un sistema conjugado de electrones π |
|
32 |
Reducción de anillos aromáticos Se utilizan soluciones de sodio o litio en amoníaco líquido, para formar compuestos alicíclicos |
|
33 |
Reducción de alquinos por el diborano En esta reacción se forman las olefinas cis |
|
34 |
Reducción de Benkeser |
|

























La reacción más característica de los alquenos es la adición al doble enlace con formación de compuestos saturados. En el doble enlace se encuentran electrones arriba y abajo del plano de éste. Durante las reacciones con reactivos electrofílicos estos electrones π se emplean para la formación de enlaces σ sin que se afecte el enlace σ original del alqueno.
La oxidación de un alqueno con diferentes reactivos se utiliza para preparar muchos compuestos y algunas veces conduce a la ruptura del doble enlace. La deshidrogenación de los ciclohexenos da compuestos bencénicos con buenos rendimientos. Finalmente los alquenos sufren algunas reacciones de sustitución en condiciones especiales, propicias a la formación de radicales libres.
No. |
REACCIóN |
EJEMPLOS |
35 |
Hidrogenación catalítica. |
|
36 |
Adición de halógenos. sólo para Cl y Br. |
|
37 |
Adición de ácido sulfúrico. |
|
38 |
Adición de HX. X = F, Cl, Br, I o bien OSO3H. |
|
39 |
Hidrohalogenación |
|
40 |
Hidratación. |
|
41 |
�Formación de halohidrinas (X2+H2O). |
|
42 |
Dimerización. |
|
43 |
Alquilación. |
|
44 |
Hidroboración. |
|
Hidroboración reductora. |
|
|
�Hidroboración oxidante. |
|
|
45 |
Oximercuriación-desmercuriación. |
|
46 |
Adición de radicales libres |
|
47 |
Adición de carbenos. |
|
48 |
Polimerización vinílica por radicales libres. |
|
49 |
Hidroxilación. |
|
50 |
Halogenación, sustitución alílica Facilidad de separación de H: alilo>3�>2�>1�>CH4>vinilo. Estabilidad de los radicales libres: alilo>3�>2�>1�>CH3>vinilo. NBS se emplea para bromar por radicales libres. |
|
51 |
Reacciones de los alcadienos El nucleófilo puede ser : X, OH, OSO3H |
|
52 |
Ozonólisis. |
|
53 |
Reacciones de polimerización: |
|
1,1-Dicloroetileno |
|
|
Cloruro de vinilo |
|
|
Tetrafluoroetileno |
|
|
Acrilonitrilo |
|
|
Cloropreno |
|
|
54 |
Reacción de Paterno-Büchi |
|
55 |
Proceso de Levinstein |
|
56 |
Hydroxilación de Milas |
|
57 |
Reacción de Sakurai |
|
58 |
Reacción de Prevost |
|
59 |
Reactivo de Normant |
|
60 |
Alilación de Trost |
|

































Métodos de preparación de los alquinos.
Puesto que el acetileno es una materia prima para varias síntesis su fabricación se produce principalmente por el tratamiento del carburo de calcio con agua.
El triple enlace se puede formar: a) sustrayendo dos moléculas de� haluro de hidrógeno de un dihaluro� adecuado o b) sustrayendo una molécula de haluro de de hidrógeno de un haluro insaturado.
Un método también valioso para algunos alquinos, es reemplazar un átomo de hidrógeno acetilénico o ambos por un metal alcalino, el cual puede ser sustituido después por otro grupo alquilo
Algunos métodos que son de interés práctico para preparar otros alquinos son:
No. |
REACCIONES |
EJEMPLOS |
61 |
Deshidrohalogenación de dihaluros de alquilo |
|
62 |
Deshidrohalogenación de monohaluros insaturados |
|
63 |
Acoplamiento oxidativos de los alquinos |
|
64 |
Alquilación de los derivados del acetileno con los metales alcalinos |
|
65 |
Acoplamiento de un alquino con un haloalquino |
|
66 |
Tratamiento del carburo de calcio con agua |
|
67 |
Adición de acetiluros a cetonas y aldehídos |
|
68 |
Adición de acetiluros a epóxidos |
|





Reacciones de los alquinos.
Los átomos de hidrógeno del acetileno (o de una alquino terminal) son bastante ácido y pueden ser sustraídos como protones por una base fuerte. Muchas de sus reacciones se basa en esta sustitución del hidrógeno por un metal alcalino y el resto de reacciones están referidas a las reacciones de adición al triple enlace.
El hecho de que los acetiluros sean buenos nucleófilo, es aprovechado para una serie de reacciones denominadas síntesis acetilénica.
No |
REACCIONES |
EJEMPLOS |
69 |
Adición de halógenos Donde X2 Cl2, Br2, en su mayoría da adición trans |
|
70 |
Adición de XH. Orientación: Obedece la regla de Markovnikov |
|
71 |
Adición de hidrógeno. |
|
72 |
Adición de agua. Hidratación. |
|
73 |
Formación de sales de metales pesados |
|
74 |
Formación de acetiluros metálicos |
|
75 |
Oxidación |
|
76 |
Hidratación catalizada por el ión mercúrico Permite obtener cetonas, tanto con alquinos terminales como internos |
|
77 |
Hidroboración -oxidación de alquinos terminales. Se utiliza un dialquil-borano impedido para evitar la adición de dos moléculas de borano en el triple enlace. El Di(sec-isoamilo)borano, denominado Disiamilborano (Sia2BH). El producto es anti-Markovnikov |
|
78 |
Reacción de Pauson-Khand |
|
79 |
Adición de alcoholes |
|
80 |
Adición de H-CN |
|
81 |
Reacción de Diels-Alder |
|
82 |
Acoplamiento de Glaser |
|
83 |
Reacción de Whiting |
|
84 |
Ciclación de Nazarov |
|
















DERIVADOS HALOGENADOS ALIFÁTICOS
Métodos de preparación
Los haluros alifáticos y alicíclicos se preparan fácilmente a partir de los alcoholes o por adición de haluro de hidrógeno a los alquenos o alquinos, siempre y cuando la forma de adición pueda predecirse. Los yoduros de alquilo se preparan frecuentemente a partir de los cloruros o bromuros por intercambio del halógeno utilizando yoduro de sodio. La brotación alílica permite introducir un átomo de bromo en un alqueno sin alterar el doble enlace.
Los siguientes son los métodos más utilizados.
No. |
REACCIONES |
EJEMPLOS |
85 |
Sustitución de un grupo oxhidrilo (OH) de los alcoholes alifáticos. |
|
86 |
Sustitución nucleofílica Grupos salientes: -OSO2R, -OSO2OR, CF3SO3- (Tf), ArSO3-, MeArSO2- (Ts), BrArSO2- (Bs) MeSO2- (Ms). |
|
87 |
Bromilación alílica |
|
88 |
Intercambio del átomo de cloro o bromo por yodo |
|
89 |
La reacción del haloformo Se puede aprovechar esta reacción de degradación, porque el principal subproducto puede tratarse del cloroformo, bromoformo o yodoformo |
|
90 |
Adición de haluros de hidrógeno a un alqueno o alquino |
Estas reacciones se explicaron en el estudio de la reactividad de alquenos y alquinos |
91 |
Adición de halógeno a un alqueno o alquino |
Estas reacciones se explicaron en el estudio de la reactividad de alquenos y alquinos |





Reactividad de los haluros alifáticos:
Los halogenuros orgánicos son intermediarios útiles en la síntesis de una gran variedad de moléculas, por lo que es muy importante conocer cómo varía la reactividad de los mismos con la naturaleza del grupo al que está unido y también con los diferentes halógenos.
Así son comunes las reacciones de sustitución nucleofílica de los halógenos, las reacciones de eliminación de los pares HX o XX que dan origen a una gran variedad de alquenos o alquinos, las reacciones de acoplamiento o formación de pares (Corey House y Wurtz).
Igualmente es de suyo importante su reactividad frente a los metales alcalinos y otros elementos pesados, que originan una química preparativa de los organometálicos que amplían el horizonte de formación de nuevas moléculas orgánicas.
Por las razones anteriores es importante repasar las preparaciones estudiadas en las secciones correspondientes.
No. |
REACCIONES |
EJEMPLOS |
92 |
Reacciones de sustitución nucleofílica Se debe tomar en cuenta: La existencia de dos mecanismos claros SN1 y SN2, con estereoquímicas definidas y la competencia entre éstas, así como con las reacciones de eliminación. |
El efecto del nucleófilo como atacante y la naturaleza del grupo saliente. El efecto del solvente |
93 |
Reacciones de eliminación Deshidrohalogenación Deshalogenación |
|
94 |
Preparación de reactivos de Grignard. |
|
95 |
Reducción. |
|
96 |
Acoplamiento o formación de pares Corey - House Reacción de Wurtz |
|



Métodos de preparación
Los alcoholes constituyen uno de los grupos de compuestos más versátiles que han sido estudiados por los químicos orgánicos. Los alcoholes pueden ser sintetizados por una variedad de reacciones relativamente directas a partir de alquenos, haluros de alquino, aldehídos, cetonas ácidos carboxílicos y varios de sus derivados. Un conjunto de reacciones sobre la preparación de los alcoholes han sido recogidas de la extensa literatura que sobre ellos existe.
No. |
REACCIONES |
EJEMPLOS |
97 |
Hidrólisis de haluros de alquilo: Sustitución nucleofílica: Sustitución de segundo orden: halogenuros primarios (y algunos secundarios) Sustitución de primer orden: halogenuros terciarios (y algunos secundarios) |
|
98 |
Hidroboración oxidación |
|
99 |
Síntesis de Favorskii-Babayan |
|
100 |
Hidrólisis de ésteres |
|
101 |
Hidratación de alquenos Esta es una reacción de adición electrofílica |
|
102 |
Hidroxilación de alquenos que dan dioles 1,2 |
|
103 |
Reacción de compuestos organometálicos con compuestos carbonílicos (Síntesis de Grignard.) Los sustratos también pueden ser halogenuro de ácido o éster para dar un alcohol terciario |
|
104 |
Reducción de aldehídos y cetonas La reducción parcial del grupo carbonilo da alcoholes; los aldehídos se reducen a alcoholes primarios y las cetonas a alcoholes secundarios. Se puede usar hidrógeno gaseoso con níquel Raney cono catalizador. Otros hidruros son el LiAlH4 y NaBH4� siendo el más selectivo el último Otra forma de reducción del grupo carbonilo, es la reacción de� Meerwein-Ponndorf-Verley, en la cual el agente reductor es un exceso de un alcohol en presencia de isopropóxido de aluminio |
|
105 |
Reducción de ácidos carboxílicos La reducción catalítica no produce ningún efecto en el grupo carboxilo y el único agente químico reductor efectivo es el hidruro de litio y aluminio |
|
106 |
Reducción de ésteres |
|
107 |
FORMACIóN DE GLICOLES |
|
108 |
A partir de la reducción de cetonas: Si se reduce una cetona con amalgama de magnesio, se obtiene un diol (pinacol) |
|
109 |
A partir de� alquenos. Se utilizan una serie de reactivos, para adicionarlos al doble enlace C=C Para que no se produzcan reacciones d eliminación, en el tratamiento de la halohidrina y los haluros vecinales se utiliza carbonato de sodio acuso, la cual es débilmente básica y suficiente para producir los dioles. |
|



















Reacciones de los alcoholes
No sólo la variedad de métodos de preparación de los alcoholes los hace importantes, sino que desde el punto de vista sintético, los alcoholes son valiosos intermediarios porque intervienen en un gran número de reacciones. Hay dos formas en las que el grupo -OH de un alcohol se ve afectado por otros reactivos:
a) Ruptura del enlace oxígeno-hidrógeno, acidez de los alcoholes:
![]()
b) Ruptura del enlace carbono-oxígeno, sustitución y eliminación:
![]()
No. |
REACCIONES |
EJEMPLOS |
110 |
Reacción con HX. Reactividad del HX: HI>HBr>HCl. Reactividad del ROH: alílico, bencílico> 3�>2�>1�. Podemos tener trasposiciones en R. También va con NaBr en sulfúrico. |
|
111 |
Deshidratación. Reactividad del ROH: 3�>2�>1�. |
|
112 |
Reacción con metales activos. Reactividad del ROH CH3OH>1�>2�>3�. M: Na, K, Mg, Al. |
|
113 |
Reacción con Ms y Ts. TsCl = |
|
114 |
Reacción de Chugaev. Los alcoholes pueden convertirse en olefinas a través de la pirólisis de sus alquilxantatos |
|
115 |
Formación de ésteres( esterificación de Fischer) |
|
116 |
Reacción con PX3. |
|
117 |
Formación de ésteres inorgánicos. |
|
118 |
Reducción de alcoholes para dar alcanos También puede utilizarse TsCl-piridina, después LiAlH4 |
|
119 |
Reacción de Schotten-Baumann |
|
120 |
Oxidación. Los oxidantes pueden ser: Na2Cr2O7, H2SO4 Na2Cr2O7, H2SO4/acetona: . Reactivo de Jones Piridina.CrO3.piridina : Reactivo de Collins (sólo forma aldehídos a partir de alcoholes primarios) Piridina.CrO3.HCl. clorocromato de piridinio ( para la formación de aldehídos a partir de alcoholes primarios. Con KMnO4. los alcoholes primarios, producen ácidos carboxílicos y lo alcoholes secundarios cetonas |
|
121 |
Síntesis de Williamson para formar éteres |
|
122 |
Carboxilación de Koch-Haaf |
|











Otras reacciones de los alcoholes

Reacciones de los Glicoles:
Muchas de las reacciones de los glicoles son típicas del grupo hidroxilo (-OH) y, por tanto, se asemejan a las reacciones de los alcoholes. No obstante, las reacciones que requieren la presencia de los grupos hidroxilo en átomos adyacentes son sólo para los glicoles. En esta sección abordaremos sólo la oxidación con el ácido peryódico: HIO4
Los glicoles, los α-oxialdehidos, las� α-oxicetonas y los compuestos α, β -dicarbonílicos son oxidados por ácido peryódico. Los productos que se muestran en las ecuaciones siguientes se forman además del ácido yódico, HIO3.
No. |
REACCIóN |
EJEMPLOS |
123 |
Oxidación de los glicoles Partiendo de glicoles y dependiendo de la naturaleza de los grupos R y R1, �se obtendrán aldehídos o cetonas |
|
124 |
Oxidación de α-oxialdehidos y cetonas: La reacción del HIO 4 con los glicoles alfa-oxialdehidos o cetonas es una reacción de oxidación-reducción. |
|
125 |
Oxidación de α,β-dicarbonílicos: |
|
126 |
Reacción de Kuhn-Winterstein |
|






Métodos de preparación y reactividad
La reacción de un metal con un haluro orgánico es un método conveniente para la preparación de derivados organometálicos, a partir de metales razonablemente activos como el Li, Mg y Zn. Los éteres en general constituyen un medio inerte, ligeramente polar con pares de electrones no compartidos sobre el oxígeno, en el cual los compuestos organometálicos generalmente son solubles.
Debe excluirse la humedad, el oxígeno y el CO2, porque de otra manera éstos reaccionarían con el compuesto organometálico, por lo cual se utiliza una atmósfera inerte de nitrógeno o helio.
Los compuestos organometálicos menos reactivos se preparan mejor a partir de haluros organomagnésicos y haluros metálicos
No. |
REACCIONES |
EJEMPLOS |
127 |
Reacción de RX con metales A los RMgX los conocemos como Reactivos de Grignard El orden de reactividad de los haluros es: RI>RBr> RCl>� RF Los fluoruros de alquilo no reaccionan con el Li ni con el Mg, mientras que el Hg sólo da resultados satisfactorios si se ha amalgamado con sodio. Los cloruro de alquilo, reaccionan muy lentamente con el Mg. Los halogenuros de vinilo, forman reactivos de Grignard sólo si se usa como disolvente tetrahidrofurano (THF). |
|
128 |
Reacciones mas comunes de los reactivos de Grignard. El formaldehído produce un alcohol primario con un reactivo de Grignard. Un aldehído superior, produce un alcohol secundario y una cetona, produce un alcohol terciario. En los ésteres, lactonas, halogenuros de acilo y anhídrido, generalmente reacciona una segunda molécula del reactivo de Grignard, con el complejo formado inicialmente, dando después de la hidrólisis un alcohol terciario con dos sustituyente idénticos. Sólo el éster del ácido fórmico da lugar a un alcohol secundario. |
|
129 |
Síntesis de éteres. Se trata un reactivo de Grignard y un compuesto que contenga un átomo de halógeno activo. Se añade un α - monocloroéter al reactivo de Grignard. Los cloroéteres del tipo ROCH2Cl, se preparan fácilmente pasando HCl por una mezcla fría de solución de formaldehído y un alcohol |
|
130 |
Síntesis de aldehídos: Se preparan por reacción entre un mol de formiato de etilo y un mol de Grignard. Sin embargo esta reacción se halla en competencia por la producción simultánea de alcoholes secundarios. Por eliminar este problema se sustituye el formiato por ortoformiato de etilo, que tiene buenos rendimientos de aldehído: |
|
131 |
Síntesis de cetonas: Las cetonas se preparan por reacción de cloruros de ácido con un Grignard. Para disminuir la formación de alcohol terciario, se agrega cloruro férrico y la reacción se lleva acabo a temperaturas inferiores a 20� C. El Grignard se adiciona a los nitrilos, para dar compuestos cuya hidrólisis conduce a cetiminas. La descomposición de éstas por ebullición con ácidos diluidos da origen a cetonas. Se obtiene buenos rendimientos, cuando se hace reaccionar un anhídrido con un equivalente molar de un Grignard a bajas temperaturas. |
|
132 |
Síntesis de ácidos: Se trata un reactivo de Grignard con dióxido de carbono sólido (CO2) y descomponiendo al final el complejo con ácido diluido |
|
133 |
Síntesis de ésteres: Se forma un éster haciendo reaccionar un reactivo de Grignard con un exceso de cloroformiato de etilo. Se mantienen en exceso el cloroformiato de etilo y añade éste sobre el reactivo de Grignard, a fin de evitar en lo posible la reacción del reactivo con el grupo carbetoxilo del éster carboxílico que se forma |
|
134 |
Síntesis de nitrilos: Un cianuro de alquilo se puede formar por reacción entre el cianógeno con un reactivo de Grignard. Los cianuros de alquilo se forman también, junto al cloruro de alquilo, cuando un reactivo de Grignard se añade a una solución etérea de exceso de cloruro de cianógeno, puesto que el cianuro de alquilo producido tiende a reaccionar con el reactivo de Grignard. |
Sólo el cloruro produce esta reacción y no así el yoduro y� bromuro. |
135 |
Reacciones de los organolitios: Son más reactivos que los Grignard. La ventaja de su mayor reactividad frente a los grupos carbonilo y nitrilo es que se efectúan menos reacciones secundarias y se obtienen mejores rendimientos. Los halogenuros de vinilo sustituido que difícilmente reaccionan con el Mg, lo hacen con el Li. En cetonas con gran impedimento estérico, por su menor tamaño, un alquil o aril litio puede alcanzar más fácilmente al grupo carbonilo protegido. Otra ventaja del litio es que reacciona con los cloruros de arilo en tanto que el magnesio no. Finalmente 2 moles de un organolitio reacciona con un mol de CO2, para formar un cetona simétrica. |
|
136 |
Reacciones de reactivos de Grignard y organolíticos con XM de metales menos electropositivos: Esta reacción es necesaria cuando se requiere organometálicos de Cd y Hg |
principalmente. |
137 |
Reactivos de Grignard con haluros de talio (TlX) Los reactivos de Grignard alílicos intervienen en reacciones de apareamiento dando rendimientos muy altos cuando se les trata con bromuro de talio. |
|
138 |
Síntesis de Corey - House. |
|
139 |
Reacciones de los organocúpricos. |
|
140 |
Preparación regioespecífica de arenos por la talación. Sustitución en para: La talación se lleva a cabo casi exclusivamente en la posición para de los compuestos bencénicos que tienen un solo grupo alquilo, halógeno o metoxilo y a temperatura ambiente. Se atribuye este comportamiento al volumen 143estérico del electrófilo. Los compuesto del para-bromobenceno pueden prepararse utilizando trifluoroacetato de talio como ácido de Lewis en las reacciones de bromación. El electrófilo es un complejo del trifluoroacetato de bromo-talio y reacciona más rápidamente en la posición para que esté más despejada. El para acetilanisol puede prepararse en forma similar utilizando trifluoroacetato de talio y cloruro de acetilo. Sustitución en orto: En todas las reacciones que puedan ocurrir en presencia de los grupos G, se formará el derivado orto-sustituido. Cada compuesto (G) contiene un oxígeno que puede formar un complejo con el trifluoroacetato de talio y fijarlo en la posición orto. Ya se sabe que este grupo de talio puede ser desplazado fácilmente con cualquier nucleófilo. Sustitución en meta: Las reacciones de talación son reversibles. La talación a temperatura ambiente se da en la posición para del compuesto bencénico. Sin embargo los bencenos talados en meta son los que predominan en la reacción. |
|
141 |
Reacciones de los organocíncicos: Los ésteres de ácidos α-halogenados al ser tratados con cinc en polvo, dan lugar a la formación de derivados organocíncicos, que se adicionan al grupo carbonilo de aldehídos y cetonas, para producir un compuesto cuya hidrólisis conduce a β-hidroxiácidos. Este procedimiento se conoce con el nombre de reacción de Reformatsky. Se emplean con resultados muy satisfactorio, los α-haloésteres de los tipos: |
|
142 |
Organometálicos con Talio: Los arenos reaccionan con trifluoroacetato de talio por sustitución aromática electrofílica. La reacción se lleva acabo generalmente en ácido trifluoroacético: La debilidad del enlace C-Tl, permite su ruptura y la consiguiente sustitución del grupo bis-trifluoroacetato tálico por diversos sustituyentes nucleofílicos |
|



























Métodos de obtención.
La mayoría de los métodos de preparación de éteres utilizan alcoholes como molécula precursora, los cuales se alquilan o arilan por pérdida de una molécula de agua a expensas de dos moléculas de un alcohol. Los alcoholes secundarios y terciarios dan rendimientos escasos por este método debido a la mayor facilidad con la cual eliminan agua en forma intramolecular para dar alqueno.
No. |
REACCIONES |
EJEMPLOS |
143 |
Deshidratación intermolecular. Es el método industrial preferido, sólo que está restringido a los éteres simétricos. |
|
144 |
Síntesis de Williamson. Mediante este método no es posible preparar ni lo éteres diarílicos ni los arilaquílicos provenientes de los fenoles correspondientes |
|
145 |
Alcoximercuriación-desmercuriación. |
|
146 |
Variantes� en la síntesis de Williamson Empleo de tosí latos: La formación del tosilato comprende sólo la ruptura del enlace O-H en el alcohol, de modo que procede con retención completa de la configuración en el carbono del alcohol. Cuando los tosí latos reaccionan con diversos nucleófilos, sale el grupo tosilato completo Empleo del sulfato de dimetilo: (CH3O)2SO2 es el éter dimetílico del ácido sulfúrico, es muy reactiva con respecto a las sustancias nucleofílicas, y cuando se emplean iones alcóxido o fenóxido, se producen los éteres metílicos correspondientes: |
|
147 |
Formación de éteres cíclicos Otra variante de la síntesis de Williamson, establece que un haluro hidroxialquil reacciona con hidróxido de sodio acuoso para generar algún ión alcóxido, que desplaza el ión haluro mediante un mecanismo SN2 del tipo intramolecular. |
|
148 |
Sustitución electrofílica en el oxígeno de alcoholes. Dado que los alcoholes no son nucleófilos fuerte, las reacciones de sustitución electrofílica directamente en el oxígeno requieren reactivos fuertemente electrofílicos tales como halógenos o iones carbonio |
|







Reactividad:
Existen pocas reacciones de los éteres debido a que el enlace C-O-C es inerte con respecto a muchos reactivos. De las pocas reacciones, se puede mencionar la relacionada de la ruptura del enlace C-O por soluciones concentradas de ácido halogenohíricos en condiciones bastante severas:
REACCIONES |
EJEMPLOS |
|
149 |
Basicidad. |
|
150 |
Escisión mediante ácidos. La reacción ocurre con un exceso de HX y calentamiento prolongado. La reactividad depende de HX: HI>HBr>HCl. |
|
151 |
Sustitución en Ca. Mecanismo radicalario. |
|
152 |
Sustitución electrófila en éteres aromáticos. OR es activante moderado La sustitución ocurre en condiciones suaves casi exclusivamente en el anillo con buenos rendimientos. El isómero para es el predominante, en algunos casos se obtiene sólo el isómero para. Existe el riesgo de que simultáneamente ocurra la ruptura del grupo éter, particularmente en la nitración y sulfonación. |
|
153 |
Éteres cíclicos. |
|
154 |
La transposición de Claisen: Los éteres fenilalílico sufren una transposición, cuando son calentados a temperaturas próximas a 200� C. El producto fenólico que se forma es el isómero orto. Pero si estuviera ocupada esta posición antes de la transposición, se obtiene el para-alilfenol. El mecanismo aceptado es aquel que indica que el grupo alilo nuca deja de estar unido al anillo durante la reacción- |
|
155 |
Reacción de Quelet |
|







Métodos de preparación:
Los métodos más comunes de preparación de los epóxidos a partir de un alqueno, que puede pasar primero por una adición de halógeno acuoso o el uso de peroxiácidos también sobre olefinas.
No. |
REACCIONES |
EJEMPLOS |
156 |
Peroxidación de dobles enlaces CC. |
|
157 |
A partir de halohidrinas |
|



Reactividad de los epóxidos:
El éter cíclico de tres miembros, el óxido de etileno, es el compuesto más simple de la familia del epóxido, los cuales desempeñan un papel especialmente importante en la química orgánica. Como se verá, son inusitadamente lábiles respecto a muchos reactivos electrofílicos y nucleofílicos.
Roseta de reacciones del epoxietano:

No. |
REACCIONES |
EJEMPLOS |
158 |
Apertura catalizada por bases �El Nu ataca al carbono menos sustituido del epóxido. |
|
159 |
Apertura catalizada por ácidos. Nu ataca al carbono más sustituido en el epóxido |
|
160 |
Reacción con reactivos de Grignard. R entra en el carbono menos sustituido del epóxido. |
|



BENCENO Y DERIVADOS
Métodos de preparación
Los alcanos C6, C7
y C8 provenientes del petróleo se pueden deshidrogenar para producir benceno,
tolueno y xilenos. A menudo el catalizador es el platino apoyado en el óxido de
aluminio (Al2O3) y los alcanos se pasan por el catalizador de
Típicamente, el petróleo sólo produce hidrocarburos, en tanto que la destilación del alquitrán de hulla da lugar (además de benceno, tolueno y xilenos) a los fenoles, cresoles y algunos hidrocarburos aromáticos polinucleares, casi como el naftaleno, antraceno y el fenantreno.
El alquitrán de hulla también produce bases heterocíclicas como la piridina y la quinolina.
No. |
REACCIONES |
EJEMPLOS |
161 |
Procesos: reformación, hidroformación y platformación La aromatización en la industria, comúnmente se llama reformación, hidroformación o platformación, y comprende una deshidrogenación-ciclación-deshidrogenación. "Faraday en 1825 obtuvo por condensación de gases de alumbrado un aceite con un olor y una fórmula empírica CH" |
|
162 |
Pirólisis de acetilenos |
|
163 |
Deshidrogenación de ciclohexenos Muchos ciclohexenos, pueden prepararse por la reacción de Diels-Alder, los que luego pueden ser aromatizados. |
|




Reactividad:
La mayor cantidad de reacciones del benceno involucran la sustitución más que la adición, lo cual se atribuye a la estabilidad del sistema del anillo aromático que se destruiría por reacciones de adición, las que son características de los alquenos y alquinos. El benceno posee una nube π rica de electrones que puede ser atacada por los electrófilos E+, razón por la cual la sustitución electrofílica es la reacción más común sobre el anillo bencénico y sus derivados, denominados "compuestos aromáticos".
No |
REACCIONES |
EJEMPLOS |
164 |
Nitración. |
|
165 |
Sulfonación. |
|
166 |
Halogenación. La sustitución electrofílica catalizada por ácidos de Lewis, tienen buenos rendimientos en el caso de la cloración y bromación Por otra parte el yodo, es tan inerte en la sustitución electrofílica, que debe utilizarse técnicas especiales para obtener derivados yodados del benceno (compuesto de sales de diazonio o talación) En cambio el flúor reacciona rápidamente con el benceno y produce una multisustitución, razón por la cual los derivados fluorurados se preparan por vía indirecta, por ejemplo, vía compuestos de� sales de diazonio o la talación. |
|
167 |
Alquilación de Friedel Crafts. Si R-X, tiene más de tres átomos de carbono, con seguridad que ocurrirán transposiciones, debido a la formación del carbocatión electrófilo más estable. Pero el carbocatión no sólo se puede obtener de los haluros de alquilo, sino que también podrían utilizarse alquenos adecuados o alcoholes, que con el reactivo adecuado formarán el electrófilo correspondiente. Restricciones: |
|
168 |
Acilación de Friedel-Crafts. Esta reacción es un medio efectivo de introducir un grupo acilo (RCO+ ) o aroilo ( ArCO+ ), en un anillo aromático. La reacción requiere de un ácido de Lewis como catalizador. El producto de la reacción es una cetona. Los cloruros de acilo y aroilo también se llaman cloruro de ácido� y se preparan fácilmente tratando ácidos carboxílicos con cloruro de tionilo (SOCl2) o pentacloruro de fósforo (PCl5). En estas reacciones no se presentan las poliacilaciones y tampoco las transposiciones. Las acilaciones, también pueden llevarse a cabo utilizando anhídrido de ácido y si éstas últimas son cíclicas, se tiene otra forma de adicionar un anillo al compuesto bencénico. |
|
169 |
Alquilación de Friedel-Crafts. En R puede haber trasposiciones. R no puede ser un Ar puesto que los ArX son poco reactivos. Esto no se produce si el benceno está sustituido con grupos que orienten a meta, o sea, atractores de electrones. Además el AlCl3 transforma a los NH, o NR en potentes atractores de electrones. Otros procesos de este tipo: |
Ar + ROH (BF3/HF/H2SO4) � Ar-R. Ar + alqueno (HF 0�C/ H3PO4) � Ar-R. |
170 |
Protonación o hidrólisis de los ácidos sulfónicos: Esta reacción se realiza con calentamiento� y con un ácido diluido (1:1). |
|
171 |
Nitrosación. |
|
172 |
�Acoplamiento diazoico. Sólo es viable en ArH muy reactivos El diazocompuesto se formar con aminas primarias mas nitrito de sodio (ácido nitroso) en medio ácido. |
|
173 |
Reacción de Bart (sustitución de un H en el benceno por el grupo arsónico) Es un buen método para introducir el arsénico en el núcleo aromático. Las sales de diazonio se tratan con arsenito de sodio en solución neutra y en presencia de sales de cobre, Co o Ni. Existen modificaciones, como la de Scheller y Hartung. |
|
174 |
Reacción de clorometilación de Blanc. Esta reacción guarda relación con la reacción de Friedel -Crafts y consiste en hacer actuar sobre el compuesto bencénico una mezcla de formaldehído y HCl en presencia de un catalizador: el cloruro de cinc, con altos rendimientos del producto. La clorometilación de los monoalquilbencenos da preferentemente derivados para. La presencia de un halógeno en el anillo bencénico dificulta la clorometilación. Los fenoles dan productos de polimerización y los grupos nitro lo inhiben. |
Pero los nitrofenoles dan buenos rendimientos. |
175 |
Reacción de Gattermann para aldehídos. Es una variación de la d Gattermann-Koch y consiste en emplear ácido cianhídrico en lugar de CO y cloruro de aluminio o cloruro de cinc como catalizadores. El cloruro cuproso no es necesario. Esta reacción es aplicable para la síntesis de. |
aldehídos fenólicos o sus ésteres, con resultados altos |
176 |
Reacción de Gattermann-Koch Los aldehídos aromáticos se pueden sintetizar mediante la reacción de una mezcla de óxido de carbono y ácido clorhídrico sobre el hidrocarburo aromático, en presencia de tricloruro de aluminio y |
cloruro cuproso como catalizadores |
177 |
Síntesis de bifenilos. �Pude llevarse a cano de varias formas, partiendo de derivados del benceno. Sin embargo el único procedimiento que se conoce a partir del benceno es el que se menciona a continuación. |
|
178 |
Arilación del benceno. Cómo no es posible utilizar un haluro aromático en la alquilación de los compuestos bencénico, el diclorometano, es una alternativa muy buena para tener dos grupos aromático unidos por un grupo |
metileno. |
179 |
Reducción de Birch Esta reacción es regioespecífica, ello se puede observar cuando en el anillo bencénico se tiene inicialmente un grupo dador de electrones (R, OH, NH2), uno de los dobles enlaces formado, involucra al carbono que tiene dicho sustituyente. En el caso de existir un sustituyente atractor de electrones (-CHO. COR. COOR, CONH2), el doble enlace no contiene al carbono en el que se halla unido dicho sustituyente. |
|
180 |
Reducción de anillos aromáticos Se utilizan soluciones de sodio o litio en amoníaco líquido, para formar compuestos alicíclicos |
|
























Dada la importancia de la alquilación y acilación de Friedel - Crafts, se hace necesario, efectuar algunas aclaraciones particularmente sobre las restricciones de esta reacción, pues es muy común en los estudiantes hacer un uso muy frecuente de estas reacciones, así como ocurre con los reactivos de Grignard.
Alquilación de Friedel- Crafts.
![]() Los haluros de arilo y vinilo no pueden utilizarse como componente haluro
porque no forman carbocationes con suficiente rapidez.
Los haluros de arilo y vinilo no pueden utilizarse como componente haluro
porque no forman carbocationes con suficiente rapidez.
![]() Es necesario cuidar las polialquilaciones. Los grupos alquilo son grupos
que liberan electrones y una vez que uno de introduce en el anillo bencénico
activa el anillo produciéndose más sustituciones( no es recomendable utilizar
un exceso del haluro de alquilo)
Es necesario cuidar las polialquilaciones. Los grupos alquilo son grupos
que liberan electrones y una vez que uno de introduce en el anillo bencénico
activa el anillo produciéndose más sustituciones( no es recomendable utilizar
un exceso del haluro de alquilo)
![]() El carbocatión que se forma de un haluro de alquilo, puede sufrir
reordenamiento, antes de atacar el anillo bencénico.
El carbocatión que se forma de un haluro de alquilo, puede sufrir
reordenamiento, antes de atacar el anillo bencénico.
![]() Las reacciones de Friedel-Crafts no se verifican cuando están presentes
en el anillo, grupos que liberan electrones en el anillo aromático o cuando el anillo tiene un grupo: -NO2, -NH2, NHR, o -NR2.
Las reacciones de Friedel-Crafts no se verifican cuando están presentes
en el anillo, grupos que liberan electrones en el anillo aromático o cuando el anillo tiene un grupo: -NO2, -NH2, NHR, o -NR2.
Acilación de Friedel - Crafts
![]() Las acilaciones no ocurren, si existe grupos desactivantes del anillo
como es el caso del grupo -NO2
Las acilaciones no ocurren, si existe grupos desactivantes del anillo
como es el caso del grupo -NO2
Grupos activantes: |
|
Poderosos: |
-NH2, -NHR, -NR2; -OH. |
Moderados: |
-OCH3, -OR. |
Débiles: |
-benceno, -CH3, -CR. |
Grupos desactivantes: |
|
Desactivantes |
-NO2, -N(CH3)3+, -CN, -COOH,-COOR, -SO3H, -CHO, -COR. |
Desactivantes, orto/para |
X |
ARENOS
Los hidrocarburos formados por grupos alifáticos y aromáticos, se conocen como arenos. El tolueno, el etilbenceno, el isopropilbenceno y el estireno son arenos. Estos compuestos pueden estudiarse desde dos puntos de vista. Se los considera como compuestos bencénicos con sustituyentes alquilo o alquenilo o pueden verse como alcanos y alquenos con sustituyentes fenilo.
Consiguientemente, en la sección precedente se ha visto con suficiente detalle los métodos de preparación de los compuestos bencénicos con sustituyentes alquílicos o alquenílicos.
Seguidamente se estudiarán algunas de las reacciones que se producen en la cadena lateral de los alquilbencenos. Se insiste en que será necesario considerar que estos compuestos son alcanos sustituidos con fenilo. Esto permitirá comprender que el grupo fenilo proporciona una reactividad especial a los grupos que se hallan en la cadena lateral del benceno.
No. |
REACCIONES |
EJEMPLOS |
181 |
Hidrogenación. |
|
182 |
Acoplamiento |
|
183 |
Reducción de Clemmensen. Se emplea para compuestos sensibles a las bases. Esto nos permite obtener ArR |
|
184 |
Reducción de Wolff-Kishner. Se emplea con compuestos sensibles a los ácidos. Permite la obtención de ArR. |
|
185 |
Modificación de los anteriores procesos. Lo que permite obtener un alqueno. |
|
186 |
Oxidación. La oxidación es total. Parece ser que al menos en los 2 primeros se necesita medio básico seguido de tratamiento en medio ácido. El proceso va en caliente. |
|
187 |
Sustitución en la cadena lateral. Halogenación radicalaria |
|
188 |
Reducción |
|
189 |
�Ozonólisis. |
|
190 |
Oxidación |
|
191 |
Sustitución en el anillo. SN aromática. |
|
192 |
Oxidación con HNO3 El ácido nítrico caliente oxida a un grupo metilo del o-xileno produciendo ácido 0-toluico El ácido nítrico caliento también oxida en forma selectiva al grupo isopropilo del p-isopropiltolueno formando ácido p-toluico |
|
193 |
Oxidación de cadenas laterales acílicas. Los compuestos de benceno con cadenas laterales acílicas se oxidan mediante agentes oxidantes fuertes a ácidos benzoicos. Las soluciones de bromo en hidróxido de sodio oxidan a los grupos acetilo a ácido |
carboxílico, y no afectan a los alquilos presentes |
195 |
Reacción de Prins |
|
194 |
Acoplamiento de Castro-Stephens |
|













Otras reacciones de interés.

Métodos de preparación:
Los fenoles se preparan generalmente a partir de los ácidos sulfónicos o de las sales de diazonio, pero a veces es conveniente preparar naftoles a partir de naftilaminas por la reacción de Bucherer.
Los métodos de preparación de mayor uso para los fenoles son los siguientes:
No |
REACCIONES |
EJEMPLOS |
196 |
Fusión del ácido bencensulfónico o sus sulfonatos con un álcali. Este método tiene la limitante de que en el anillo aromático no deben encontrarse grupos sensibles a los medios básicos o a las condiciones extremas de |
calentamiento. |
197 |
A partir de halobencenos. Un método importante fue creado por la compañía química Dow, que consisten en hacer reaccionar el clorobenceno y el hidróxido de sodio acuoso a una temperatura y presión elevadas. La mezcla de la. |
reacción se debe acidular con un ácido mineral fuerte, como el HCl o el H2SO4, para obtener el fenol. |
198 |
Hidrólisis de ésteres |
|
199 |
Sustitución de un grupo diazonio por OH- El grupo diazonio puede sustituirse con un grupo oxhidrilo simplemente acidificando y calentando mucho la mezcla reaccionante. |
|
200 |
Naftoles a partir de naftilaminas. �La conversión reversible entre los naftoles y naftilaminas, se denomina reacción de Bucherer. La naftilamina se calienta con una solución acuosa de bisulfito de sodio o ácido sulfúrico. |
|
201 |
Por sustitución nucleofílica aromática (SN) Esto se da si el haluro bencílico cuenta además con otros sustituyentes desactivadores del mismo. |
|
202 |
Fenoles a partir de reactivos organotalio Los fenoles pueden prepararse a partir de los ditrifluoroacetato de aril talio por reacción con tetraacetato de plomo y trifenilfosfina. |
|
203 |
A través de la formación de peróxidos. |
|
204 |
Reacción de Wolffenstein-Boters |
|







Reactividad de los fenoles:
Los fenoles se caracterizan por la formación de soluciones coloreadas al agregar cloruro férrico. Los fenoles sufren sustituciones electrofílicas en las posiciones orto y para con mayor facilidad, debido al efecto de resonancia del par de electrones libres en el átomo de oxígeno. Sin embargo, muchas de las reacciones comunes de los alcoholes no se aplican a los fenoles.
El efecto orientador del grupo oxidrilo en las posiciones orto es muy notable. Una muestra de ello son las reacciones de Orbe-Schmitt y la de Reimer-Tiemann.
Cuando el fenol reacciona con el formaldehído, se produce una reacción de polimerización, para producir la baquelita.
No. |
REACCIONES |
EJEMPLOS |
205 |
Reacciones de acidez. Los fenoles son más ácido que los alcoholes, pero menos ácidos que los ácidos carboxílicos, por esa razón forman sales fácilmente al combinarse con bases. En vez de un haluro es preferente el uso de sulfato de dimetilo |
|
206 |
Síntesis de Williamson. En la formación de los aliléteres, no se olvide que se presenta la transposición de Claisen al ser calentado el éter alílico, para dar un fenol orto sustituido o para de esta previamente ocupada la posición orto. |
|
207 |
La reacción d e Gattermann. Los derivados aldehídicos de fenoles en posición para son preparados por la reacción de Gattermann, utilizando cloruro� cianuro de cinc, como catalizador y benceno como solvente |
|
208 |
Formación de ésteres. |
|
209 |
Hidrogenación: Se produce en derivados bencénicos que tienen un sustituyente activador del benceno |
|
210 |
Reacción de Reimer - Tiemann. Consiste en la introducción del grupo aldehído en los fenoles en las posiciones orto y para, con el predominio de los orto fenólicos, por tratamiento de fenoles con cloroformo en presencia de álcalis. La presencia de grupos desactivadores del anillo (-NO2, CN, COOH o SO3H) que orientan a meta, hace bajar el rendimiento drásticamente. Si una de las posiciones orto estuviese ocupada, el grupo aldehído ingresa a la posición para. |
|
211 |
Reacción de Kolbe - Schmitt. Carbonatación Se denomina así a la síntesis de ácidos fenol carboxílicos, por calentamiento de fenóxidos alcalinos con dióxido de carbono a 4� 8 atm de presión. La reacción es aplicable a otros fenoles y generalmente se verifica con buenos rendimientos. La carboxilación suele producirse en orto con el fenol, pero también puede realizarse en para, si se emplea fenóxido de potasio. La naturaleza del metal alcalino influye sobre la orientación del grupo carboxilo. A temperaturas inferiores a 200� C lo anterior información funciona, pero a temperaturas mayores a 200�, el único isómero que se obtiene es el para sin importar el tipo de metal alcalino. |
|
212 |
Reordenamiento de Bamberger |
|
213 |
La reacción de Duff. Permite obtener exclusivamente aldehídos fenólicos en posición orto. Consiste en calentar fenol con hexametilentetramina (urotropina), en medio glicérico y en presencia de ácido bórico |
|
214 |
Síntesis de Bucherer |
|
215 |
Reacción de von Pechmann Esta reacción consiste en la condensación de un fenol con un β-cetoéster. Los agentes de condensación son el á. sulfúrico,� cloruro de aluminio o el pentóxido de fósforo o ácido polifosfórico. Con una variación de los fenoles y el β-cetocarbonilo se pueden obtener una gran variedad de derivados de la cumarina. Así por ejemplo si se utiliza el resorcinol como sustrato fenólico, se obtiene otro derivado de la cumarina Si el compuesto dicetónico es el ácido málico en presencia del ácido sulfúrico se transforma en el hemialdehído malónico, que actúa en su forma tautómera, condensándose con el fenol. Con el pirogalol se forma la dafnetina |
|
216 |
Formación de Quinonas Los fenoles y aminas aromáticas, por oxidación de las mismas producen con altos rendimientos quinonas una base muy importante para la industria de los colorantes. Un sustituyente activante en l posición para en el fenol, coadyuva a la formación de quininas con mejores rendimientos |
|
217 |
Reacciones de sustitución aromática electofílica en el anillo bencénico de fenoles. Los fenoles son sustratos muy reactivos a la sustitución aromática electrofílica, porque los electrones no enlazantes del grupo hidroxilo estabilizan al complejo sigma que se forma por ataque en la posición orto o para. Los fenoles son sustratos excelentes para la halogenación, nitración, sulfonación y algunas reacciones de Friedel - Crafts. Como son muy reactivos, generalmente los fenoles se alquilan o acilan con catalizadores relativamente débiles (como el HF) para evitar sobrealquilación o sobreacilación. |
|
218 |
Nitración de Zicke |
|
219 |
Reacciones de importancia en síntesis: |
|
220 |
Reacción de Houben - Hoesch. Las cetonas fenólicas se pueden preparar mediante la síntesis de Hoesch, que consiste en condensar un polifenol en posición meta con acetonitrilo y ácido clorhídrico, en presencia de cloruro de cinc como catalizador y llevando a ebullición al final. |
|

























Métodos de preparación
Un método importante de preparación de los haluros de arilo es la sustitución aromática electrofílica directa y otro método, comprende el uso de los reactivos organotalio. Las sales de diazonio, Ar-N2+, es otro intermediario importante.
No. |
REACCIONES |
EJEMPLOS |
221 |
Halogenación directa. |
|
222 |
Halogenación con sales de diazonio |
|
223 |
Las Reacciones de Talación Una de las variantes y que no pasa por la talación del anillo, es el uso de del trifluoroacetato de talio como reactivo de Lewis, cuando el halógeno es bastante reactivo, como es el caso del bromo |
|
224 |
Reacción de Wolffenstein-Boter |
|





Reactividad:
El enlace carbono - halógeno en los haluros de arilo y vinilo, es el principal causante de la falta de reactividad hacia la sustitución nucleofílica, que sin embargo ocurre si se dan algunas condiciones estructurales en el haluro de arilo, es decir si además del halógeno en el anillo bencénico se hallan grupos sus tractores de electrones, tales como:
-SO3H -COOH -CN -NR3 -COR -CHO
NO. |
REACCIONES |
EJEMPLOS |
225 |
Formación de reactivos de organometálicos. |
|
226 |
Sustitución nucleófila aromática sustitución bimolecular. En general ArX+Z�ArZ+X-. �Z: NaNH2, NaCN, etc. En todo caso, el anillo debe contener grupos (G) fuertemente atractores de electrones, situados o/p respecto al X. |
|
227 |
Sustitución electrófila aromática directa. X es desactivante, pero dirige a orto y para. |
|
228 |
Eliminación-adición. El bencino, el reactivo atacante debe ser una base fuerte, y el anillo no estar activado para desplazamiento bimolecular. |
|
229 |
Acoplamiento de Castro-Stephens |
|
230 |
Síntesis de Rosenmund-von Braun |
|





El grupo carbonilo� >C=O debido a la polaridad de su enlace, es uno de los grupos funcionales más importantes en química orgánica y se hallan en los aldehídos, cetonas, ácidos carboxílicos y sus derivados. En esta sección se estudiará la preparación y reactividad de aldehídos y cetonas.
Método de preparación de Aldehídos.
Puesto que los aldehídos se oxidan y reducen con facilidad, los métodos y técnicas que deben emplearse para preparar aldehídos son relativamente especiales.
No. |
REACCIóN |
EJEMPLOS |
231 |
Oxidación de alcoholes primarios La oxidación con ácido crómico es el método más común de preparación en el laboratorio. (Reactivo de Collins, reactivo de Jones, etc.) |
|
232 |
Ruptura de dobles enlaces C=C por el O3 La ozonólisis puede utilizarse para preparar aldehídos o centonas, dependiendo de la estructura del alqueno de partida |
|
233 |
Por reducción selectiva de ésteres. |
|
234 |
Oxidación de metilbencenos La cloración del grupo metilo se efectúa por radicales libre y el intermediario formado se hidroliza en medio básico para formar el aldehído. |
|
235 |
Oxidación de glicoles por el ácido peryódico |
|
236 |
Reducción de cloruros de ácido El hidruro de tri-tert-butoxialuminio y litio, LiAl[OC(CH3)3]3, es un agente reductor altamente selectivo y permite transformar los haluros de acilo o bencilo a aldehídos. Reducción de Rosenmund. Los aldehídos alifáticos y aromáticos se pueden preparar a partir de los correspondientes haluros de acilo o bencilo, por una hidrogenación controlada por un catalizador de paladio envenenado con azufre o sulfato de bario |
|
237 |
Hidroboración - oxidación de de alquinos terminales. Para esto se utiliza el di(sec-isoamil) borano que se llama disiamilborano |
|
238 |
Alquilación de 1,3 ditianos. El 1,3 ditiano es un ácido débil que se puede desprotonar por medio de una base fuerte como el n-butillitio, el carbanión resultante puede desplazar el halógeno de un haluro de alquilo primario o tosilato que se hidroliza con una solución ácida de cloruro mercúrico. |
|
239 |
Síntesis de Gatterman -Koch Esta reacción tiene éxito si en el anillo existen grupos activantes del mismo para una sustitución electrofílica aromática |
|
240 |
A partir de nitrilos. |
|
241 |
Reacción de Reimer-Tiemann. Como ya se dijo en la sección correspondiente, esta reacción permite obtener aldehídos fenólicos |
|
242 |
Oxidación de RX 1� con DMSO |
|
243 |
Oxidación de alquilboranos. |
|
244 |
Ozonólisis reductora |
|
245 |
Formilación |
|
246 |
Deshidrogenación catalítica. Se pasa el vapor de alcohol a través de un tubo caliente que contiene el catalizador. R no puede ser más grande que el grupo etilo |
|
247 |
Naftaldehidos a partir de sus cloruros de acilo: |
|
248 |
Reacción de Stephen El procedimiento para la obtención de aldehídos, basado en la reducción de un nitrilo por el cloruro estannoso anhidro, disuelto en éter y saturado con ácido clorhídrico, se conoce con el nombre de reacción de Stephen (1925) |
|







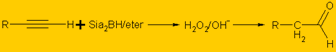









Métodos de preparación de Cetonas.
Al igual que los aldehídos, las reacciones involucradas para la obtención de las cetonas, siguen siendo oxidaciones y reducciones, siguiendo un estrecho paralelismo a la preparación de aldehídos.
No. |
REACCIONES |
EJEMPLOS |
249 |
Oxidación de ROH secundarios. |
|
250 |
Reacción de cloruros de ácido con organocúpricos. |
|
251 |
Ozonólisis de alquenos. |
|
252 |
Síntesis de cetonas a partir de ácidos carboxílicos Los reactivos de organolitio se pueden emplear para sintetizar las cetonas a partir de los ácidos carboxílicos, en una primera instancia se forma un dianión, que luego se protona para perder rápidamente agua y formar la cetona |
|
253 |
Acilación de Friedel Crafts |
|
254 |
Síntesis acetoacética |
|
255 |
Descarboxilación de ácidos b-cetónicos y malónicos. |
|
256 |
Otra posibilidad: |
|
257 |
Oxidación de alquilboranos. |
|
258 |
Hidratación de alquinos terminales |
|
259 |
A partir de 1,3-ditianos. |
|
260 |
Síntesis de cetonas a partir de nitrilos Un reactivo de Grignard o de organolito, ataca a un nitrilo para dar la sal de magnesio de una imina. La hidrólisis ácida de la imina produce la cetona. |
|
261 |
Cetonas a partir de cloruros de ácido y organocadmio. Los compuestos de Grignard son demasiado reactivos para ser utilizados en la preparación de cetonas. Sin embargo, si primero se convierte en un alquilcadmio tratándolo con cloruro de cadmio anhidro, la reacción subsecuente con un cloruro de ácido produce un buen rendimiento de cetona. |
|
262 |
Cetonas a partir de dialquilcupratos de litio Una solución etérea de dialquilcuprato de litio se trata con un cloruro de ácido a temperaturas muy bajas, el producto que se forma es una cetona. Esta es una variación de la síntesis de Corey-House |
|
263 |
Oxidación de compuestos alicíclicos. |
|
264 |
Reacción de Sugasawa |
|
265 |
Ciclación de Nazarov |
|
266 |
Acilación reductiva de Nenitzescu |
|
267 |
Reacción de Pauson-Khand |
|
268 |
Reacción de Thorpe |
|

















Reacciones de los aldehídos y cetonas
El grupo carbonilo de cetonas y aldehídos, puede sufrir reacciones de adición nucleofílica sobre el átomo de carbono del carbonilo, debido a la polaridad positiva que se da en el mismo. Por otro lado, algunos reactivos electrofílicos, pueden combinarse prioritariamente con los pares de electrones del átomo de oxígeno que conforma el grupo carbonilo. Muchas reacciones de condensación son comunes a los grupos carbonilo y por otro lado, la posición alfa al grupo carbonilo también está activada para una reacción ácido-base.
Finalmente los dobles enlaces conjugados con el grupo carbonilo de aldehído y cetonas, como también de ésteres, tienen un comportamiento especial para reacciones de adición conjugada.
No |
REACCIONES |
EJEMPLOS |
269 |
Oxidación de metil cetonas. Reacción del haloformo |
|
270 |
Oxidación de Baeyer-Williger. Facilidad de migración: H>fenilo>R 3�>R 2�>R 1�> metilo |
|
271 |
Reducción a alcoholes. |
|
272 |
Reducción a hidrocarburos. |
|
273 |
Reacción de Wittig. |
|
274 |
Oxidación. |
|
275 |
Adición de reactivos de Grignard. |
|
276 |
Reacciones de ciclación con hidrazina Cuando la acroleína reacciona con hidrazina, el producto es un dihidropirazol |
|
277 |
Adición de CN, formación de cianohidrínas. |
|
278 |
Formación de iminas y enaminas R, necesariamente a de ser a |
|
279 |
Aminación reductiva. |
|
280 |
Adición de derivados del amoníaco. Los aldehídos y las cetonas reaccionan con varios derivados de amoniaco |
|
281 |
Típicamente, la reacción se continúa en medio ácido: |
|
282 |
Formación de tioacetales y tiocetales. |
|
283 |
Halogenación de cetonas. |
|
284 |
Reacción de Cannizzaro. El proceso sólo tiene lugar con aldehídos sin Ha. Se obtiene la sal correspondiente y un ROH. |
|
285 |
Reacción de Canizzaro cruzada. Necesariamente uno de los dos aldehídos debe ser formaldehído |
|
286 |
Ciclaciones por condensaciones aldol. |
|
287 |
Condensación aldólica (la adición de iones enolato a aldehídos y cetonas): En la condensación aldólica se une el Ca de uno de los compuestos al C carbonílico del otro. Podemos reducir el grupo carbonilo a aldehídos y cetonasa, b insaturadas |
limpiamente con LiAlH4 Otro reductor útil es el NaBH4 que nos permite reducir el doble enlace. |
288 |
Anelación de Robinsón (formación de anillos). La secuencia de una adición conjugada seguida de una condensación aldólica simple para adicionar un anillo a otro, se conoce como la anelación de Robinsón. |
|
289 |
Condensación aldólica cruzada. Esto sólo tiene utilidad sintética cuando uno de los compuestos carece de Ha y consecuentemente no puede autocondensarse. En todos los demás casos se obtienen mezclas más o menos complejas. |
|
290 |
Un esquema completo de la utilidad de la condensación aldólica es: |
|
291 |
Reacción de Mannich. Los compuestos que contienen un átomo de hidrógeno activo se condensan con amoniaco, o con aminas primarias, o secundarias (generalmente en forma de clorhidrato), y formaldehído. |
El átomo de hidrógeno activo puede provenir de un grupo metileno activado por la vecindad de un grupo cetónico, nitroalcanos, etc. |
292 |
Racemización en Ca. |
|
293 |
Adición conjugada de reactivos de Cu. También con CH3Cu y luego con agua. |
|
294 |
Reacción de Claisen-Schmidt. Los aldehído aromáticos se condensan, por acción de los álcalis, con aldehídos alifáticos o con cetonas, originando aldehídos o cetonas no saturados en alfa, beta. El requisito es que la cetona o aldehído tenga un grupo metileno activo, para generar el carbanión en el medio básico de la reacción En 1929 KUHN introdujo una extensión de la reacción de Claisen al condensar en condiciones bien determinadas el aldehído |
Crotónico con otro aldehído, mejoró los rendimientos utilizando acetato de piperidina como catalizador. |
295 |
Condensación con nitroalcanos. La reacción de Claisen-Schmidt, también se amplia a la reacción del benzaldehido con compuestos con metileno activo |
|
296 |
Condensación de Stobe Los aldehídos o centonas en presencia de alcóxidos, se combinan con los ésteres succínicos, produciendo los ácidos dibásicos no saturados. Los agentes de condensación básicos utilizados son el ter-butóxido potásico y el hidruro de sodio |
|
297 |
Condensación de Darzens La reacción que comprende la condensación de un aldehído o cetona con éster α-halogenado, con producción de un α,β-epoxiéster, es conocida con el nombre de condensación del éster glicídico de Darzens. El agente de condensación puede ser etóxido de sodio o amiduro de sodio. La saponificación de los ésteres glicídico, seguida de acidificación, conduce a |
epoxiácidos que luego forman una acetona o un aldehído por descarboxilación, seguida de� transposición |
298 |
Reacción de Perkin Consiste en condensar un aldehído aromático con un anhídrido alifático, en presencia de sal sódica o potásica del correspondiente ácido carboxílico. Se calienta el sistema porque se utiliza una base débil. El producto es un ácido cinámico |
Cuando hay presente un oxhidrilo en orto, se cierra el anillo con la cadena ácida no saturada. |
299 |
Condensación con nitrilos. |
|
300 |
Condensación de Jhonson. Jhonson ha mejorado el rendimiento de la condensación de Stobe, utilizando ter-butóxido potásico en ter-butanol. El producto de condensación� es tratado con HBr en caliente, formándose una γ-lactona, que es abierta por NaOH y luego reducida con� H2/Cu-CrO3 como catalizador. Se utiliza este método para obtener compuestos policíclicos. |
|
301 |
Reacción de Reilly Las metilencetonas están sujetas a ataque oxidativo específico en las posiciones α. La oxidación de Reilly con SeO2 conduce a α-dicetonas. |
|
302 |
Reacción de Wittig Se conoce con el nombre de reacción de Wittig la reacción de trifenilfosfina-alquilideno con un compuesto carbonílico de aldehído o cetona, para formar una olefina, en que el átomo de oxígeno |
carbonílico es reemplazado por un grupo metileno |
303 |
Adición a aldehídos y cetonas a-b insaturados |
|
304 |
Compuestos dicetónicos a partir de las metiléncetonas El ácido nitroso y sus ésteres convierten las metilen cetonas en α.cetoximas, que al ser tratadas por solución acuosa de HCl, libera el compuesto dicetónico |
|
305 |
Reagrupamiento de Baeyer-Villiger |
|
306 |
Reactivo de Normant |
|
307 |
Reacción de Stork |
|
308 |
Reacción de Paterno-Büchi |
|
309 |
Síntesis de Boord |
|
310 |
Reacción de Scholl |
|
311 |
Reacción de Bradsher |
|
312 |
Reacción de Haller-Bauer |
|
313 |
Reacción de Allan-Robinson |
|


















































Reacciones de los cetenos (CETENAS)
|
El ceteno CH2=C=O, es el miembro más simple de un grupo de compuestos que llevan el mismo nombre. El ceteno gaseoso (p.e. -41�) es muy reactivo especialmente en presencia de nucleófilos. El ceteno se obtiene industrialmente por pirólisis de la acetona: CH3COCH3 |

NITRILOS
Método de Obtención.
Los nitrilos no se pueden preparar fácilmente a partir de los ácidos carboxílicos, pero un ácido sulfónico se transforma en el nitrilo por calentamiento con un cianuro inorgánico, la deshidratación de una amida, la sustitución de haluros o aromático mediante la reacción de Sandmeyer o la cianoetilación, son métodos que generalmente dan buenos rendimientos.
No. |
REACCIONES |
EJEMPLOS |
314 |
A partir de haluros de alquilo RX(1�). |
|
315 |
A partir de aldehídos o cetonas (cianohidrinas) |
|
316 |
Reducción de amidas. |
|
317 |
Reacción de Sandmeyer |
|
318 |
Deshidratación de amidas.Cuando las amidas se calientan con pentóxido de fósforo, cloruro de tionilo o una mezcla de cloruro de aluminio y cloruro de sodio o simplemente con anhídrido acético hirviente, se presenta una deshidratación de la amida produciendo un nitrilo. |
|
319 |
CianoetilaciónSi en la adición de Michael, se utiliza como sustrato el acrilonitrilo, se origina un� proceso denominado cianoetilación, el cual es un método para sustituir un hidrógeno activo por el grupo -CH2CH2CN. Un hidrógeno activo o sustituible puede estar unido a un átomo de carbono, nitrógeno u oxígeno a condición de que preferiblemente haya un� sólo hidrógeno activo. |
|
320 |
Sustitución de un grupo sulfónicoLos ácidos sulfónicos experimentan una sustitución nucleofílica del grupo ácido en condiciones drásticas, así el grupo ácido sulfónico puede remplazarse por un grupo oxhidrilo, ciano o amino. Los nitrilos que con mejor rendimiento se obtienen son los que utilizan ácidos sulfónicos policíclicos. |
|
321 |
Talación.El grupo bis-trifluoroacetato de talio puede sustituirse con un grupo ciano en dos formas distintas: por reacción de los bis-trifluoroacetato de aril talio con cianuro cuproso en dimetil formamida (DMF) o por reacción con cianuro de potasio acuoso bajo la acción de la luz ultravioleta |
|






Reactividad de los nitrilos:
Los nitrilos son considerados derivados de ácido porque se hidrolizan fácilmente para formar primero amidas y posteriormente ácidos carboxílicos, con el mismo número de carbonos que el nitrilo original.
No. |
REACCIONES |
EJEMPLOS |
322 |
Adiciones nucleofilicas. |
|
323 |
Reducción de nitrilos |
|
324 |
Obtención de amidas |
|
325 |
Adiciones electrofílicas |
|
326 |
Obtención de aldehídos y cetonas. |
|
327 |
Obtención de ésteres. |
|
328 |
Obtención de cetonas |
|
329 |
Reacción de Ehrlich-Sachs |
|
330 |
Reacción de Houben-Hoesch |
|







Métodos de preparación de las Aminas.
No. |
REACCIONES |
EJEMPLOS |
331 |
Reducción de nitro compuestos. |
|
332 |
Básicamente para aminas aromáticas. Podemos reducir selectivamente un determinado grupo NO2 tratándolo con SH2 en NH3 |
|
333 |
Reacción de RX con amoníaco o aminas. Lo mismo para el caso de los ArX, pero en este caso Ar debe tener sustituyentes con tendencia a atraer electrones. En realidad se obtiene la sal, de la que obtenemos la amina por tratamiento en medio básico: |
|
334 |
A partir de derivados alifáticos nitrados. |
|
335 |
Aminación reductiva. |
|
336 |
Síntesis de Gabriel, exclusivo para obtener aminas primarias |
|
337 |
Reducción de nitrilos, oximas y amidas |
|
338 |
Degradación de amidas alifáticas y aromáticas, según Hoffmann. |
|
339 |
Basicidad de las aminas, como característica importante para comprender su reactividad |
|
340 |
Alquilación. Las aminas alquiladas son más reactivas que la amina original, por lo que tienden a la multialquilación |
|
341 |
Empleo en síntesis. |
|
342 |
Sustitución anular en aminas aromáticas. Los grupos -NH2, -NHR y -NR2 son activantes poderosos dirigiendo a o/p. |
El grupo NHCOR es activante menos poderoso que -NH2 |
343 |
Conversión a amidas. Las aminas terciarias no se convierten en amidas. |
|
344 |
Eliminación de Hofmann en sales de amonio cuaternarias. |
|
345 |
Reacciones con ácido nitroso. |
|
346 |
Reacción de Bechamp |
|
347 |
Reagrupamiento de Hoffmann-Martius |
|
348 |
Reacción de Sugasawa |
|
349 |
Reordenamiento de Bamberger |
|
350 |
Reagrupamiento de Reilly-Hickinbotton |
|
351 |
Reacción de Dakin-West |
|
352 |
Reacción de Buchwald-Hartwig) |
|























Métodos de obtención
Las aminas primarias alifáticas reaccionan con ácido nitroso mediante una reacción llamada diazoación formando sales de diazonio alifáticas, muy inestables. A temperatura ambiente esta sales se descomponen perdiendo nitrógeno y formando carbocationes que reaccionan produciendo mezclas de alquenos, alcoholes y haluros de alquilo.
Las aminas
aromáticas primarias reaccionan con ácido nitroso formando sales de aril
diazonio. Aún siendo las sales de aril diazonio inestables, son mucho más
estables que las sales de diazonio alifáticas y no se descomponen, cuando la
temperatura de la reacción se mantiene a menos o igual a
![]()
Consiguientemente el proceso debe ocurrir a temperatura baja y utilizarse el reactivo formado inmediatamente (in situ).
Reactividad.
Las sales de aril diazonio son intermediarios sintéticos muy útiles en la química de los compuestos aromáticos, porque el grupo diazonio puede ser sustituido por muchos otros grupos, incluyendo:
-F, -Cl, -Br, -I, -Cn, -OH y -H. La reacción de Sandmeyer es uno de los mejores ejemplos.
Debido a su inestabilidad, no es necesario aislarlo, razón por la cual se agregan a la reacción a bajas temperaturas reactivos como CuCl. CuBr, KI,et, para lograr la sustitución correspondiente.
Sólo en la sustitución del grupo diazonio con -F es necesario aislar una sal de diazonio. Esto se hace adicionando HBF4 a la mezcla reaccionante, para precipitar el fluoroborato de diazonio, ArN2+ BF4- que es soluble y estable.
Por otro lado las sales de diazonio son electrófilo débiles, reaccionan con compuesto aromáticos muy reactivos produciendo compuesto azo.
Esta reacción de sustitución aromática electrofílica se conoce con frecuencia como reacción de copulación diazo.
No. |
REACCIóN |
EJEMPLOS |
353 |
Reemplazo del H |
|
354 |
�Reemplazo por -H. |
|
355 |
Reacción de Sandmeyer. |
|
356 |
�Reemplazo por -I. |
|
357 |
�Reemplazo por -F. |
|
358 |
�Reemplazo por -OH. |
|
359 |
�Copulación diazoica. |
G: debe ser un grupo con fuerte tendencia a liberar electrones: OH, NR2, NHR o NH2 |





La preparación de un colorante azoico, comprende las reacciones de diazoación y diazocopulación (o simplemente copulación). Todas las moléculas precursoras de copulación utilizadas para obtener colorantes azoicos, poseen un carácter común, esto es: un átomo activo de hidrógeno ligado a un átomo de carbono, consiguientemente los sustratos, pueden ser:
![]() Compuestos que poseen hidróxido fenólicos: fenoles y naftoles
Compuestos que poseen hidróxido fenólicos: fenoles y naftoles
![]() Aminas aromáticas
Aminas aromáticas
![]() Moléculas con grupo cetónico enolizable de carácter alifático
Moléculas con grupo cetónico enolizable de carácter alifático
![]() Moléculas heterocíclicas : pirrol, indol, etc.
Moléculas heterocíclicas : pirrol, indol, etc.
ACIDOS CARBOXíLICOS
Métodos de obtención
Los ácidos carboxílicos son uno de los compuestos orgánicos más importantes y representan un estado de oxidación avanzado del átomo de carbono, varios de sus métodos de preparación involucran otros grupos funcionales, que en las anteriores secciones han sido debidamente estudiadas
Sin embargo y con la finalidad de sistematizar las mismas, indicaremos entre muchas otras reacciones que producen ácido, las siguientes, que resultan ser los mejores métodos de preparación:
No, |
REACCIONES |
EJEMPLOS |
360 |
Oxidación de alquenos con KMnO4 |
|
361 |
Por oxidación de aldehídos y alcoholes primarios |
|
362 |
Por oxidación de alquilbencenos |
|
363 |
Por oxidación de metilcetonas |
|
364 |
Por hidrólisis de cianhidrinas y otros nitrilos |
|
365 |
Por carbonatación de los reactivos de Grignard. Este método se aplica a los haluros primarios, secundario, terciarios, de alilo, bencilo y arilo. |
|
366 |
La síntesis malónica |
|
367 |
La reacción de Kolbe. Es una reacción específica para formar ácido salicílico |
|
368 |
Reacción de von Richter |
|








Reactividad de los ácidos carboxílicos:
Los ácidos carboxílicos contienen también al grupo carbonilo, pero sus reacciones son muy diferentes de las de las cetonas y aldehído, estas últimas, reaccionan normalmente por adición nucleofílica de del grupo carbonilo, pero los ácidos carboxílicos (y sus derivados) reaccionan principalmente por sustitución nucleofílica de acilo, donde un nucleófilo sustituye a otro en el átomo de carbono del acilo (C=O).
No. |
REACCIONES |
EJEMPLOS |
369 |
Formación de sales Todos lo ácidos pueden ser neutralizados con las diversas bases inorgánica, para producir sales orgánicas |
|
370 |
Formación de cloruros de ácido Los mejores reactivos para convertir los ácidos carboxílicos en cloruros de ácido, son el cloruro de tionilo (SOCl2) y el cloruro de oxalilo (COCl)2, porque forman subproductos gaseosos que no contaminan al producto. El cloruro de oxalilo hierve a 62� Cy se evapora de la mezcla de reacción. |
|
371 |
Esterificación de Fischer La reacción del ácido con un alcohol catalizado por ácido, recibe el nombre de esterificación de Fischer. El ácido protona el grupo carbonilo y lo activa hacia el ataque nucleofílico del alcohol. |
|
372 |
Esterificación con diazometano Se pueden convertir los ácidos carboxílicos en sus ésteres metílicos de una manera sencilla, agregando una solución de diazometano en éter. |
El subproducto es el nitrógeno gaseoso y el diazometano en exceso se evapora... |
373 |
Condensación de ácidos con aminas. Síntesis directa de amidas La reacción inicial de un ácido carboxílico con
una amina da una sal de carboxilato de amonio. Calentando esta sal a
temperaturas mayores a
|
|
374 |
Reducción de los ácidos carboxílicos a alcoholes. El hidruro de litio y aluminio reduce los ácidos carboxílicos a alcoholes primarios. El diborano, también reduce selectivamente los carbonilos de los ácidos carboxílicos antes que cualquier otro carbonilo (cetonas, aldehídos, etc.) al correspondiente alcohol primario. |
|
375 |
Reducción a aldehídos Esta reacción ocurre a partir del derivado clorado del ácido carboxílico y se utiliza un agente reductor muy suave como el tri(t-butoxi)hidruro de litio y aluminio, |
LiAl[OC(CH3)3]3H. Bajo estas condiciones el cloruro de ácido se reduce a aldehído que se aísla posteriormente. |
376 |
Formación de cetonas. El ácido carboxílico reacciona con dos equivalentes de un reactivo de organolitio. El primer equivalente del organolitio desprotona al ácido. El segundo se agrega al carbonilo para formar un dianión |
estable. La hidrólisis del dianión, forma el hidrato de una cetona |
377 |
Reacción de Hunsdiecker En esta reacción el ácido carboxílico es tratado con una base de un metal pesado, como Ag2O, HgO, o Pb(OAc)4 para formar la sal del metal pesado. A continuación se agregan bromo o yodo y se calienta la mezcla de reacción. Se forma el halogenuro del metal y un halogenuro del compuesto orgánico con |
pérdida de un átomo de carbono. |
378 |
Reacción de Hell-Volhard-Zelinsky (VHZ). El ácido es tratado con bromo molecular catalizado por fósforo rojo o PBr3, resultando |
Que un hidrógeno del carbono α de ácido es sustituido por el bromo, para producir un α-bromo ácido. |
379 |
Reacciones de descarboxilación Los ácidos carboxílicos aromáticos experimentan la descarboxilación al fundirse la sal de sodio del ácido con cal sodada (NaOH + CaO). Los ácidos alifáticos reaccionan en condiciones más vigorosas, con el riesgo de llegar a descomponerse. |
Estas reacciones tienen bajo rendimiento, por lo que su uso se halla en estudios de degradación. |
380 |
Descarboxilación de ácidos dicarboxílicos Dos tipos de ácidos dicarboxílicos experimentan la descarboxilación simplemente al calentar el ácido libre. El ácido etanodioico (ácido oxálico) sufre la pérdida de CO2 para producir el ácido fórmico. El ácido propanodioico (ácido malónico) y los ácidos propanodioicos sustituidos |
pierden CO2 y dan el ácido acético o un acético sustituido como producto |
381 |
Conversión de un α-haloácido: Un α. haloácido, puede sufrir una sustitución nucleofílica del halógeno y transformarse en una serie de derivados del ácido que por otra vía sería imposible de obtenerlos: Conversión en un ácido dicarboxílico Conversión en un α-oxiácido Conversión en un α-aminoácido Conversión en un ácido α,β-insaturado |
|
382 |
Reacción de Schmidt Los ácidos carboxílicos, en presencia de ácido sulfúrico concentrado, pueden ser |
convertidos en aminas primarias con un átomo de carbono menos mediante el ácido hidrazoico, para dar aminas |
383 |
Reacción de Henke |
|
384 |
Reacción de Ugi |
|
385 |
Reacción de� Rosenmund |
|
386 |
Conversión a anhídrido |
|
















DERIVADOS DE Ácidos CARBOXíLICOS
CLORUROS DE ACILO
Métodos de preparación.
Aún cuando se conocen otros haluros de acilo, los cloruros son los más versátiles para su manejo en el laboratorio. Existe una analogía de sustitución del -OH de alcoholes por los reactivos que también sustituyen el -OH de los ácidos carboxílico. Sin embargo no es posible formar un haluro de ácido con los HX, pero si éste último puede reemplazar a un -OH de un alcohol. De manera que puede escribirse una forma general de preparación de los haluros de ácido, concretamente de los cloruros:

No. |
REACCIONES |
EJEMPLOS |
387 |
Formación del cloruro de ácido |
|

Reactividad de los cloruros de ácido
Los cloruros de acilo son intermediarios importantes en la preparación de los ácidos carboxílicos y derivados de los mismos.
No. |
REACCIONES |
EJEMPLOS |
388 |
Formación de amidas Cuando un cloruro de acilo reacciona con un exceso de amoniaco acuoso concentrado y frío, el cual se obtiene como hidróxido de amonio concentrado, se produce una amida, ésta es una reacción típica de sustitución acilnucleofílica. El amoníaco puede sustituirse por aminas primarias o secundarias, las terciarias no pueden usarse, porque no posen un hidrógeno activo. |
|
389 |
Formación de ésteres Cuando un cloruro de acilo aromático es uno de los reactivos, se agrega una base para eliminar el HCl a medida que se forma. La base puede ser una solución de NaOH diluido, trietilamina o piridina, en estos últimos casos el procedimiento se conoce como el método de Schotten-Baumann. Si se emplea un fenol en lugar de un alcohol, la reacción procede con la formación de un éster arílico. |
|
390 |
Hidrólisis de los cloruros de acilo Los cloruros de acilo se hidrolizan al calentarse con agua, pero la reacción es acelerada por la adición de una base (como NaOH). Los cloruros de ácido aromático reaccionan mucho más lentamente que los alifáticos |
|
391 |
Formación de anhídridos Los cloruros de acilo reaccionan con el anión carboxilato, RCOO-, que es neutralizada para obtener el ácido libre. Sirve para preparar anhídridos asimétricos preferentemente. |
|







ÉSTERES
Métodos de preparación:
Uno de los métodos más comunes de preparación de ésteres, es la reacción de un cloruro de acilo y un alcohol o fenol. Es más común la reacción de Fischer, que contempla la reacción de un ácido y un alcohol catalizada por ácidos fuertes.
El mecanismo de la esterificación es la más estudiada, por lo que se puede afirmar que se conoce muchos aspectos de las mismas, en función de las variables que intervienen para la formación de un éster.
No. |
REACCIONES |
EJEMPLOS |
392 |
Esterifiación de Fischer Este método es una reacción de equilibrio y es particularmente versátil debido a que la mayoría de los alcoholes pueden conseguirse en el mercado y son poco costosos. El alcohol a menudo sirve como disolvente de la reacción. El equilibrio se desplaza hacia la formación del producto por separación permanente del agua formada, por diferentes métodos |
|
393 |
Reacción del anión carboxilato con un haluro de alquilo Esta reacción SN2 se limita a los haluros de alquilo metilo, primarios y secundarios, ya que el anión carboxilato presenta algunas propiedades básicas y promueve la eliminación en haluros terciarios. |
|
394 |
Reacción de alcoholes con anhídridos Los acetatos se forman por esta vía, debido a la facilidad de formación del anhídrido acético, igualmente se usa ampliamente en la acetilación de carbohidratos y celulosa |
|
395 |
Preparación de ésteres metílicos mediante el uso del diazometano (AR)R-COOH +� CH2N2 →� (Ar)R-COOCH3 + N2 |
|
396 |
γ-y δ - Oxiácidos: Formación de Lactonas Estos compuestos contienen el gripo -OH y -COOH, razón por la cual espontáneamente o bajo la influencia del calor, formen ésteres cíclicos denominados lactonas. Por esa misma razón, las lactonas son abiertas por diversos nucleófilos como el agua, amoníaco o un alcohol. Se ha observado que las lactonas se forman siempre que sea posible estructurar un anillo de cuatro (β - lactonas) cinco o seis átomos, algunas veces en compuestos que ya contienen un anillo. Las lactonas suelen producirse internamente cuando se genera un anión carboxilato en una molécula que contiene átomo de halógeno. Esta es una reacción típica� SN2, la cual requiere que la geometría de la molécula muestre que el ión carboxilato y el halógeno s encuentran en las distancias adecuadas, para cerrar el éster, formando la lactona. |
γ-oxiácido γ-lactona δ - oxiácido δ - lactona |









Reactividad de los ésteres:
Generalmente los ésteres presentan reacciones mediante el mecanismo de sustitución acilnucleofílica
No. |
REACCIONES |
EJEMPLOS |
397 |
Hidrólisis: La hidrólisis de ésteres puede llevarse a cabo en condiciones ácidas o básicas. En medio ácido la reacción es reversible, en cambio en medio básico no, |
debido a la formación de una sal carboxílica. (saponificación) |
398 |
Con Amoniaco. Amonólisis El amoniaco� es el nucleófilo que ataca al átomo de cabono acilo deficiente en electrones, después de lo cual ocurre la sustitución alnucleofílica, para producir una amida. |
|
399 |
Con los reactivos de Grignard Un exceso de reactivo de Grignard actúa sobre el éster en condicones anhidras, seguido por la hidrólisis para producir un alcohol. El alcohol producido de esta manera siempre contiene dos grupos Ar o R proporcionados por el reactivo de Grignard.. Cuando el sustrato es el carbonato de dietilo, con un exceso de reactivo de Grignard se introducen tres sustituyentes idénticos en el carbono del acilo. El carbonato de dietilo se prepara fácilmente a partir del fosgeno con 2 moles de etanol y en medio básico: Cl.CO-Cl + EtOH → EtO-CO-OEt |
|
400 |
Transesterificación: Los ésteres reaccionan con otros alcoholes en condiciones ácidas o básicas para producir nuevos ésteres y esta reacción se onoce como transesterificación ya que un éster se transforma en otro. La transesterificaión es ampliamente usada en la preparación de la fibra sintética conocida como Dacrón, un poliéster preparado a partir del tereftalato de dietilo o dimetilo y el etilenflicol |
|
401 |
Con agentes reductores. Reducción. La hidrogenación de un éster es más difícil de lograr que la hidrogenación de un alqueno, alquino, aldehido o cetona. Requiere de condiciones extremas de temperatura y presión y un catalizador cromito de cobre (Cu2O y Cr2O3). Por tal razón es preferible en el laboratorio, recurrir al hidruro de litio y aluminio en medio de éter anhidro y el otro, sodio en etanol. El hidruro de litio tiene buenos rendimientos y ambos métodos no atacan dobles o triples enlaces C-C aislados. Elmétodo del sodio metálico en etanol se conoce como procedimiento Bouveault-Blanc |
|





AMIDAS
Métodos de preparación:
Los métodos más comunes para preparar amidas, RCONH2, o amidas N-sustituídas, RCONHR y RCONRR,, utilizan un sustrato acílico y aminas o amoniaco como nucleófilos.
No. |
REACCIONES |
EJEMPLOS |
402 |
A partir de cloruros de acilo y aminas El método de laboratorio más versatil para prearar amidas no sustituidas como sustituídas, ocurre entre un cloruro de acilo y amoniaco o la amina adecuada. Pueden prepararse amidas alifáticas o aromáticas. |
|
403 |
A partir de ácidos y amoniaco Cunado se trata un ácido con un exceso de amoniaco, se forma la sal de amonio del ácido. Después, la sal de amonio se calienta vigorosamente para formar la amida correspondiente y una molécula de agua. |
|
404 |
A partir de ésteres y amoniaco La mayoría de los ésteres tanto alifático como arílicos, reaccionan con amoníaco para formar la amida correspondiente. |
|
405 |
A partir de anhídridos y amoniaco Este tipo de reacción es de utilidad comercial, sólo cuando se dispone fácilmente del anhídrido necesario. |
|



Reacciones de las amidas
Las reacciones más comunes de las amidas siguen el patrón general de la sustitución acilnucleofílica.
No. |
REACCIONES |
EJEMPLOS |
406 |
Hidrólisis de las amidas Las amidas pueden hidrolizarse en condiciones ácidas o básicas, hasta el ácido carboxílico correspondiente o su sal como productos respectivos. |
|
407 |
Deshidratación de las amidas: Formación de nitrilos. Las amidas se convierten suavemente y con buenos rendimientos a los nitrilos correspondientes, al calentarse con SOCl2, PCl5, P2O5 o POCl3 (oxicloruro de fósforo) como el más indicado. Esta reacción es la mejor alternativa, porque los haluros de alquilo secundarios y/o terciarios no reacciona en condicones SN2 con unión cinuro. |
|
408 |
Otras reacciones de las amidas: La conversión de una amida a una amina que contiene el mismo número de átomos de carbono y la conversión de una amida a una amina primaria que contiene un átomo de carbono menos (reacción de Hofmann) |
|

ANHíDRIDOS
Métodos de síntesis
No. |
REACCIONES |
EJEMPLOS |
|
409 |
A partir de los ácidos monocarboxílicos El anhídrido de los monoácidos más importantes es el anhídrido acético, que se prepara por deshidratación del ácido acético, para formar inicialmente un compuesto intermediario, denominado cetena (o ceteno), que luego se combina con otra molécula de ácido acético, para producir el anhídrido respectivo: |
La cetena también puede prepararse por la pirólisis de la acetona a 700� (CH3COCH3 → CH2=C=O + CH4). |
|
410 |
A partir de anhídridos y ácidos carboxílicos Para formar otros anhídridos, se puede hacer uso de un exceso del anhídrido acético y dos moles de ácido respectivo, originando otro anhídrido simétrico con |
buenos rendimientos,.el ácido acético que se forma se elimina por destilación |
|
411 |
A partir de los ácidos dicarboxílicos Los ácidos oxálico y malónico no producen anhídridos por calentamiento, en cambio los ácidos succínico y glutárico, si lo hacen porque tienen la opción de formar anhídridos cíclicos de cinco y seis miembros respectivamente. El ácido adípico (seis carbonos) y otros diácidos alifáticos de cadena recta superiores no forman anhídridos El ácido ftálico forma anhídridos por calentamiento y no así los ácidos isoftálicos y tereftálico, por otro lado también el ácido maleico, forma un anhídrido y no así el |
|
|
ácido fumárico, debido a que sus grupos carboxilo se. hallan en una posición trans, que no permite la deshidratación |
El isómero ácido fumárico no reacciona |
||






Reacciones de los anhídridos:
El tipo de reacción más general que experimentan los anhídridos es la apertura de la unión anhídrido con el uso de un nucleófilo adecuado agua, amoniaco, alcohol, etc.). Los anhídridos cíclicos se ven afectados de igual manera. En este último caso, pueden originarse alternativas de síntesis muy interesantes para generar algunas estructuras que por otras vías son muy difíciles de obtenerlos.
No. |
REACCIONES |
EJEMPLOS |
412 |
Reacción con reactivos nucleofílicos: agua, alcohol y amoniaco La adición de trazas de ácido mineral o ión oxidrilo al agua aumenta la velocidad de la hidrólisis considerablemente, y también la reacción de alcohólisis es agilizada por la adición d de trazas de ácido mineral o un ión alcóxido al alcohol. En los anhídridos cíclicos en el extremo no sustituido, contiene el grupo ácido carboxílico cuyo OH puede ser sustituido por un haluro. |
|
413 |
Síntesis de Haworth |
|
414 |
Formación de cetonas ( reacciones de Friedel - Crafts) Los anhídridos pueden emplearse en la acilación de Friedel-Crafts de compuestos aromáticos Los anhídridos cíclicos a su vez dan la oportunidad de formar anillos condensados luego de dos acilaciones seguidas de F-C. |
|





ADICIONES Y CONDENSACIONES DE LOS ENOLES E IONES ENOLATO
Formación y reactividad de los enoles e iones enolato
Muchas de las reacciones ya se han visto en las secciones correspondientes y solo ejemplificaremos aquellas reacciones que no fueron aún consideradas o en su caso aquellas cuyo uso es importante en la síntesis orgánica.
Las reacciones más útiles de los iones enolato y sus intermediarios implican el ataque a los grupos carbonilo. Esas reacciones pueden dar adición nucleofílica al carbonilo (cetonas y aldehídos) o sustitución nucleofílica de acilo (derivados de ácido, especialmente ésteres).
No. |
REACCIONES |
EJEMPLOS |
ALDEHIDOS Y CETONAS |
||
415 |
Halogenación alfa-promovida por bases La halogenación promovida por una base se efectúa por un ataque nucleofílico de un ión enolato en la molécula electrofílica del halógeno. Los productos son la cetona halogenada y un ión halogenuro. Debido a la tendencia a la halogenación múltiple (prueba del haloformo), se prefiere la catálisis ácida. |
Donde X = Cl, Br y I |
416 |
Halogenación alfa-catalizada con ácido. Implica el ataque de la forma enólica de la cetona a la molécula electrofílica de halógeno. En esta reacción se puede reemplazar un hidrógeno alfa o más empleando las cantidades adecuadas de halógeno. A diferencia de las cetonas, los aldehídos se oxidan con facilidad y los halógenos son agentes muy oxidantes, de ahí que los |
intentos de halogenar un aldehído, culminaron en su oxidación a ácidos carboxílicos. |
417 |
Reacción de Hell-Volhard-Zelinsky El ácido se trata con bromo y tribromuro de fósforo y a continuación se agrega agua para hidrolizar al bromuro de alfa-bromo acilo. |
|
418 |
Condensación aldólica de los aldehídos y cetonas. La condensación aldólica implica la adición nucleofílica de un ión enolato a otro grupo carbonilo. El producto, que es una beta-hidroxicetona o aldehido, se llama aldol. Bajo las condiciones adecuadas, el aldol se |
Esta reacción sufre catálisis ácida y/o básica. puede deshidratar para formar un compuesto carbonílico alfa,beta-insaturado. |
419 |
Condensación aldólica promovida por base. Bajo condiciones básicas, la condensación aldólica se presenta por la adición nucleofílica del ión enolato (nucleófilo fuerte) a un grupo carbonilo. |
La protonación forma un aldol. |
420 |
Condensación aldólica catalizada por ácido El enol funciona como nucleófilo débil para atacar a un grupo carbonilo activado (protonado). |
|
421 |
Deshidratación de los productos de Aldol. Al calentar una mezcla ácida o básica que contenga un aldol, se efectúa la deshidratación de la función alcohol. El producto es un aldehído o cetona alfa, beta - insaturados. |
|
422 |
Ciclaciones aldólicas. Frecuentemente se forman anillos de cinco y seis miembros por reacciones aldólicas intramoleculares de las dicetonas. Las ciclaciones aldólicas de anillos de más de seis miembros y menores de cinco, son menos comunes, debido a que su formación está menos favorecida. En algunos casos un grupo carbonilo puede quedar fuera del anillo formado en el producto. |
|
423 |
Alquilación de iones enolato Un ión enolato puede funcionar como nucleófilo, alquilándose en el proceso. Como el enolato tiene dos sitios nucleofílicos (el oxígeno y el carbono alfa), puede reaccionar en cualquiera de ellos. Por lo general la reacción se efectúa, principalmente en el carbono alfa, formando un nuevo enlace C-C.. |
Las bases y alcóxidos provocan reacciones secundarias, por lo que se hace uso del DAL: [(i-Pr)2N-Li+)]. Diisopropil amiduro de litio. Los aldehídos no se prestan para esta alquilación, porque tienden a sufrir reacciones secundarias con el DAL. |
424 |
Formación y Alquilación de enaminas Una alternativa más suave para dirigir la alquilación de los iones enolato es la formación y alquilación de un derivado de enamina. Una enamina (amina vinílica) es el análogo nitrogenado de un enol. Y se forma en la reacción de una cetona o aldehído con una amina secundaria. Una enamina es un nucleófilo más fuerte que un enol, pero todavía es muy selectivo en las reacciones de alquilación. |
Algunos halogenuros que reacciona bien con enaminas son los siguientes: Ph-CH2-X, >C=C-CH2-X, CH3-X, RCO-Cl Halogenuros halogenuro halogenuros halogenuros de bencilo de alilo de metilo de acilo |
425 |
Los aldehídos que no contienen hidrógeno en la posición alfa no pueden formar iones enolato, consiguientemente no participan en la condensación aldólica, a menos que reaccione con el ión enolato de otro compuesto carbonílico (reacción aldólica cruzada). En cambio cuando un aldehído de este tipo ( o dos aldehídos diferentes) es sometido a la acción de una base fuerte, ocurre una oxidación-reducción de la misma molécula (de las moléculas diferentes) llamada reacción de Cannizzaro. |
Esta reacción tiene utilidad limitada por el número de aldehidos sin hidrógenos alfa existentes. |
426 |
La condensación aldólica convierte un C=O en C=C, pero es limitada. Una nueva formar de originar un doble enlace carbono-carbono a partir de un grupo carbonilo y un carbanión, es la reacción denominada de Wittig., donde un carbanión estabilizado por fósforo (iluro) ataca una cetona o aldehído. Una eliminación subsecuente produce un alqueno. |
|
ÉSTERES |
||
427 |
Condensación de Claisen. Los hidrógenos alfa de los ésteres son débilmente ácido y pueden eliminarse por la acción de una base para dar iones enolato. Los ésteres son menos ácidos que las cetonas y aldehídos. La condensación de Claisen resulta cuando una molécula de éster sufre la sustitución nucleofílica de acilo por un enolato de éster. El intermediario tiene un grupo alcoxi (-OR) que actúa como grupo saliente, formando un beta-cetoéster. |
|
428 |
Condensación de Dieckman Es una ciclización de Claisen, es decir es una condensación intramolecular. |
|
429 |
Condensaciones de Claisen Cruzadas Las condensaciones de Claisen se pueden efectuar entre ésteres diferentes, en especial cuando sólo uno de ellos tiene los hidrógenos alfa necesarios para formar un enolato. Un éster sin hidrógenos alfa funciona como componente electrofílico (benzoatos, formiatos, carbonatos y |
oxalatos). |
430 |
Condensación de Darzens |
|
431 |
Condensaciones de los ésteres y las cetonas. Las condensaciones de Claisen cruzadas, también son posibles entre cetonas y ésteres. Las cetonas son más ácidas que los ésteres y se acepta que el enolato de la cetona sea el nucleófilo en la condensación. Los rendimientos son mayores si el éster no tiene hidrógenos alfa. Sin embargo es posible la condensación en el caso de que ambos tengan hidrógenos alfa, pues la mayor acidez de la cetona, proporciona el ión enolato. |
|
432 |
Síntesis de Kiliani-Fischer |
|
433 |
Condensación de Stobbe Los ésteres succínicos reaccionan con aldehídos o cetonas en presencia de alcóxidos, dando los ácidos dibásicos no saturados. Los agentes de condensación más utilizados son, el ter-butóxido potásico y el hidruro de sodio |
|
434 |
Condensación de Perkin Los anhídridos también experimentan reacciones de condensación en presencia de una base, esta última suele ser la sal del carboxilato correspondiente al ácido en el anhídrido, en lugar de las bases alcóxidas anteriormente utilizadas. Este tipo de reacción recibe el nombre de condensación de Perkin. |
|
435 |
Grupos Nitro y Ciano como sustituyentes acidificantes El grupo ciano (-CN) y el grupo nitro (-NO2) también sustraen electrones y hacen que los hidrógenos alfa, sean un tanto ácidos. Los carbaniones alfa resultantes, son estabilizados por resonancia. Como consecuencia de ello este tipo de compuestos experimentan la condensación con los aldehídos (y algunas cetonas) que). |
no contienen ningún hidrógeno α (como el benzaldehido |





























COMPUESTOS β-DICARBONíLICOS
Existen diversas combinaciones posibles de aldehídos, cetonas y ésteres que pueden clasificarse como compuestos β-dicarbonílicos, en esta sección no centraremos en las beta-dicetonas, los beta-cetoésteres y los beta-diésteres, con las siguientes estructuras generales:
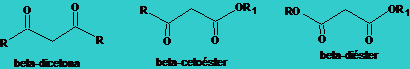
No, |
REACCIóN |
EJEMPLOS |
436 |
Condensación de Knoevenagel Esta reacción es considerada como una ampliación a la de Perkin. La condensación de aldehídos y cetonas con un grupo metileno activo en presencia de una base orgánica se llama reacción de Knoevenagel. |
Los compuestos con metileno activo, pueden ser, el éster malónico, el éster acetilacético, el cianato de etilo. |
437 |
SíNTESIS MALóNICA Síntesis del malonato de dietilo: |
|
438 |
Reacciones de acilación Para la acilación se cambia la base, ya que el etóxido en alcohol reaccionaría primero con el haluro de acilo. Se sustituye por sodio metálico o hidruro de sodio, que forman sales del malonato respectivo |
|
439 |
Reacciones de alquilación La secuencia de pasos que se inicia con el éster malónico y que produce un derivado del ácido mono o disustituido, se conoce como la síntesis del éster malónico . Los dos pasos claves son 1) la alquilación del éster malónico y 2) la hidrólisis y descarboxilación del éster malónico sustituido. Podría también utilizarse algunos haluros de arilo como : Ph(CH2)n-X, donde n ≥ 1 |
|
440 |
SíNTESIS ACETOACÉTICA Síntesis del acetoacetato de etilo |
|
441 |
Reacciones de alquilación. |
|
442 |
Reacciones de Acilación Las consideraciones efectuada para la síntesis malónica son perfectamente aplicables para la síntesis acetoacética |
|








COMPUESTOS DE CARBONILO α, β - INSATURADOS
En esta sección se toman en cuenta reacciones significativas de adición de los reactivos nucleofílicos a los compuestos de carbonilos α, β- insaturados (aldehídos, cetonas y ésteres)
La adición de un reactivo polar, H-Nu, a un compuesto carbonílico α, β- insaturado puede ocurrir de una de las dos maneras siguientes:
Adición -1,2. Adición al doble enlace carbono-oxígeno
Adición-1,4 o adición conjugada, donde el nucleófilo se adiciona al doble enlace conjugado con el grupo carbonilo.
No- |
REACCIONES |
EJEMPLOS |
||||
443 |
Adición de los reactivos de Grignard a aldehídos conjugados con un doble enlace. El grupo aldehído es más reactivo con respecto a la adición nucleofílica que el grupo ceto, razón por la cual reaccionan predominantemente por la adición-1,2. y está favorecida si los grupos R tanto en el aldehído como en el reactivo de Grignard son pequeños. |
Sin embargo, cuando el Grignard es voluminosos como en el caso del bromuro de tert-butilmagnesio, el principal modo de adición es la adición 1,4 o conjugada. |
||||
444 |
Adición de los reactivos de Grignard a cetonas conjugados con un doble enlace. Estas cetonas tienen menos tendencia a experimentar la adición 1,2 debido a la menor reactividad del grupo ceto a la adición nucleofílica. La cantidad de adición 1,2 disminuye aún más cuando el sustituyente unido al grupo carbonilo es voluminoso. |
La presencia de sustituyente en la posición 4 en un sistema conjugado, reduce la cantidad de producto de adición-1,4 ya que restringen el ataque del reactivo de Grignard. |
||||
445 |
Reacción de Adición de MICHAEL Los compuestos con grupos metilénicos activos (tales como cianacetato de etilo, fenilacetato de etilo, cianuro de bencilo, acetilacetato de etilo, etc.) dan productos de adición con cetonas y ésteres α,β no saturados. La reacción se cataliza con bases, como etóxido de sodio, piperidina, etc . Por otro lado esta reacción es reversible Reaccionantes en la adición conjugada
|
|
||||
446 |
Cianoetilación: Cuando en la reacción de Michael el sustrato es el acrilonitrilo, se tiene un proceso muy útil que se conoce como cianoetilación, el cual es un método para sustituir un hidrógeno activo por el grupo -CH2CH2CH.. El hidrógeno activo o sustituible puede estar unido a un átomo de carbono, nitrógeno u oxígeno, pero es preferible que haya un solo hidrógeno activo, pues de otro manera podrían entrar más de un grupo cianoetilo. El valor sintético de esta reacción radica en gran parte por las reacciones que puede sufrir el grupo nitrilo, como ya se vio en la sección correspondiente. |
|
||||
447 |
Alilación de Trost) |
|
||||
448 |
Síntesis de Meyers |
|
||||
449 |
Reacción de Franchimont |
|
||||
450 |
Reacción de Baylis-Hillman |
|
||||
















Otras reacciones de interés
No. |
REACCIóN |
EJEMPLO |
451 |
Reacción de Prins |
|
452 |
Acoplamiento de Suzuki) |
|
453 |
Acoplamiento de Glaser |
|
454 |
Alilación de Trost |
|
455 |
Reacción de Pschorr |
|
456 |
Reacción de Twichell) |
|
457 |
Catalizador de Adkins |
|
458 |
Reacción de Ferrario |
|
459 |
Reagrupamiento de Amadori |
|
460 |
Ciclación de Nazarov |
|
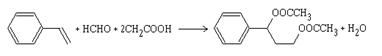










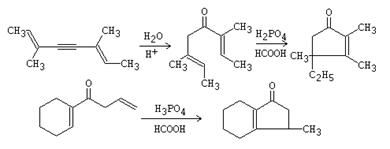
Bibliografía
- CASON, J. "Química Orgánica Moderna". Edit. URMO S.A. España. 1975
- ELLIS, G.P. . "Química Orgánica". Edit.
- GUEVARA J.D. ET AL. "Química de las Reacciones Orgánicas". Edit. Alhamabra S.A. Madrid 196
- HORN y STRAUSS. "Problemas de Química Orgánica". Edit. Noriega-Limusa. México. 1988
- MORRISON Y BOYD. "Química Orgánica". Edit. Addison-Wealwy Iberoamericana. Quinta Edición. 1990
- QUIORED. Recursos Educativos de Química Orgánica. www. quiored.
- REUSCH, W.H. "Química Orgánica". Edit. -McGraw-Hill. México D.F. 1979
- SOLOMONS, T.W.G. . "Química Orgánica". Edit. Limusa-México.. 1988
- WADE L.G., JR. "química Orgánica". Edit. Préntice-Hall Hispanoamericana, S.A. -Segunda Edición. 1993
- WINGROVE - CARET. "Química Orgánica", Edit. Harla México 1984
Wilbert Rivera Muñoz
Licenciado en Ciencias Químicas
Especialista
en Didáctica de
Máster en Educación Superior
Docente
Titular de Química Orgánica en
| Este apunte fue enviado por su autor en formato DOC (Word).
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. |
Comentarios de los usuarios
Agregar un comentario:
Aún no hay comentarios para este recurso.
Boletín de Novedades

Acceso rápido
• Monografias
• Examenes
• Enlaces
• Publicar material o sitio
• Foros
• Diversion
• Test de Orientacion
• Institutos
• Secundarios
• Universidades de Argentina
• Residencias Universitarias
• Guia de Carreras Universitarias y Cursos
• Cultura del Mundo
• Buscador en tu sitio


Sobre ALIPSO.COM
Monografias, Exámenes, Universidades, Terciarios, Carreras, Cursos, Donde Estudiar, Que Estudiar y más: Desde 1999 brindamos a los estudiantes y docentes un lugar para publicar contenido educativo y nutrirse del conocimiento.
Contacto »Legales